

Химическая физика, 2022, T. 41, № 12, стр. 81-89
Механизм многостадийного захвата N2O5 на покрытии из метановой сажи
В. В. Зеленов 1, *, Е. В. Апарина 1
1 Федеральный исследовательский центр химической физики им. Н.Н. Семёнова Российской академии наук
Москва, Россия
* E-mail: v.zelenov48@gmail.com
Поступила в редакцию 28.01.2022
После доработки 16.02.2022
Принята к публикации 21.02.2022
- EDN: DEGRYK
- DOI: 10.31857/S0207401X22120111
Аннотация
Исследован захват N2O5 на сажевом покрытии при T = 255 и 298 К в диапазоне концентраций [N2O5] = 1.3 ⋅ 1012–3.3 ⋅ 1013 cм–3 с использованием проточного реактора с подвижной вставкой и масс-спектрометрической регистрацией. Получена серия зависимостей коэффициента захвата от времени экспозиции покрытия к газу-реагенту в указанном диапазоне концентраций. В рамках ленгмюровского представления адсорбции предложен механизм многостадийного захвата, который объясняет сложную временнýю зависимость коэффициента захвата и форму его зависимости от концентрации. На базе предложенного механизма из аналитического описания экспериментальных данных получен ряд элементарных параметров, позволяющих моделировать величину коэффициента захвата при произвольных концентрациях N2O5 и температуре.
ВВЕДЕНИЕ
Окислы азота NOx играют немаловажную роль в физико-химических процессах земной атмосферы. Один из тропосферных окислителей, а именно N2O5, образуется по реакциям NO2 + O3 → NO3 + + O2 и NO3 + NO2 ↔ N2O5. Вторая реакция является обратимой, при этом константа равновесия смещается в сторону образования N2O5 при понижении температуры. Натурные измерения относительных концентраций N2O5 вблизи поверхности дают следующие их значения: 50–100 ppt (1 ppt = 10–12) над Арктикой в зимний период и до 3.8 ppb (1 ppb = 10–9) над загрязненными регионами континентов [1]. Основными путями стока N2O5 являются: прямой (гидролиз на капельках облаков) и непрямой, связанный с убылью NO2 или NO3, а также захват N2O5 на твердом атмосферном аэрозоле различного состава [1].
Средняя масса аэрозоля в тропосфере составляет 4.6 мкг · м–3. При этом на долю органической фракции приходится около 27%, большей частью которой является углерод [2]. Глобальная эмиссия углерода составляет ∼9 Тг в год [3], а средняя концентрация субмикронных частиц углерода в воздухе оценивается в 0.6 мкг · м–3 [4, 5]. В основном углеродный аэрозоль образуется в результате функционирования различных технических устройств, в которых используется горение топлив, а также от сельскохозяйственной деятельности и лесных пожаров.
Данные лабораторных измерений реакционного захвата N2O5 на ряде неорганических субстратов, имитирующих тропосферный аэрозоль, показали, что эффективность таких процессов зависит от природы аэрозольных частиц, влажности (RH) и температуры. При захвате N2O5 на компонентах морской соли и других модельных субстратах значения коэффициентов захвата γ находятся в пределах 0.02–0.0002; для черного углерода γ ≈ 0.005 [6]. Коэффициент захвата зависит от концентрации газофазного реагента и растет с увеличением относительной влажности [1, 7].
В литературе имеется всего несколько работ по исследованию захвата N2O5 на покрытии из сажи ряда углеводородов, а также на частичках элементного углерода с использованием различных методик [8–12].
В работе [8] исследовался захват N2O5 на покрытии из частиц древесного угля. В отсутствие воды наблюдалась физсорбция с последующей окислительно-восстановительной реакцией и образованием NO в качестве продукта. С увеличением влажности возрастает роль гидролиза с образованием азотной кислоты на поверхности.
В работе [9] исследовали захват N2O5 на покрытии из сажи горения метана и пропана. Установлена временная зависимость коэффициента захвата с его начальным значением γ0 = 0.016 (в расчете на геометрическую поверхность) и стационарной величиной γss = 6 · 10–3 спустя 10 мин. С использованием смоговой камеры в условиях, близких к естественным, исследован захват N2O5 в присутствии NO2 на покрытии из электроискровой сажи при относительной влажности RH = = 50%. Из анализа временнóго профиля продуктов сделана оценка коэффициента захвата N2O5: γ = 4 · 10–5, с образованием азотной кислоты, не зависящая от времени взаимодействия.
В работе [11] исследовали захват N2O5 на покрытии из сажи горения гексана. Установлена временная зависимость коэффициента захвата с его начальным значением γ0 = 0.1 и стационарной величиной γss = 3 · 10–2 при [N2O5] = 1 · 1012 cм–3 в расчете на геометрическую поверхность. Установлено, что коэффициент стационарного захвата γss обратно пропорционален концентрации N2O5 при [N2O5] < 3 · 1012 cм–3 и не зависит от [N2O5] выше этой пороговой концентрации.
В работе [12] исследован времязависимый захват N2O5 на покрытии из метановой сажи в диапазоне концентраций газа-реагента 1 ⋅ 1012–3 ⋅ 1013 cм–3. Сделана оценка соотношения реакционной и адсорбционной составляющих в общем процессе захвата. В качестве единственного газофазного продукта захвата зарегистрирован NO. При этом образование NO количественно соответствует ∼60% израсходованного N2O5. По форме зависимости коэффициента начального захвата от концентрации N2O5 сделан вывод о механизме захвата через последовательность элементарных стадий, включающих обратимую адсорбцию, образование поверхностного комплекса и его последующий мономолекулярный распад.
Цель данной работы состояла в поиске аналитического представления коэффициента захвата N2O5 на покрытии из свежей метановой сажи во всем временнóм диапазоне процесса захвата, начиная от его начального и до стационарного значения. Это позволит количественно предсказывать эволюцию процесса захвата N2O5 аэрозольными сажевыми частицами в реальных условиях тропосферы при произвольных температурах и концентрациях N2O5 и использовать эти данные в современных математических моделях захвата газов-реагентов на поверхности жидкостей и твердых тел [13–17].
ЭКСПЕРИМЕНТ
Химический реактор. Захват N2O5 исследовали в проточном реакторе с подвижной вставкой и нанесенным на нее пленочным сажевым покрытием [18]. Реактор сопряжен с масс-спектрометром высокого разрешения с электронной ионизацией. Диапазон энергии ионизирующих электронов можно варьировать от 50 до нескольких эВ при разбросе в 0.1 эВ. Ионизация при пониженной энергии электронов позволяет идентифицировать продукты реакции на масс-спектральных линиях, которые при рутинном масс-спектральном анализе при 70 эВ являются суперпозицией масс-спектральных пиков как исходного газа-реагента, так и продуктов.
Основной поток гелия (чистоты 99.99%) протекает через термостатированную ампулу, заполненную кусочками тефлоновых капилляров и намороженной на них N2O5, а затем – через цилиндрический стеклянный реактор внутренним диаметром dR = 1.3 см с линейной скоростью u = = 80–260 см · с–1 при суммарном давлении в реакторе p = 1.3–3.0 Торр и регулируемый вентиль откачки. Все внутренние поверхности реактора и вентилей покрыты тефлоновой пленкой марки Teflon FEP. Тонкий центральный стержень из нержавеющей стали внешним диаметром dr = 0.2 см и максимальной длиной L = 50 см с сажевым покрытием можно перемещать вдоль оси трубки реактора из компенсирующего объема в зону контакта с газом-реагентом с помощью внешнего магнита. Через этот компенсирующий объем подается дополнительный поток гелия, во избежание неконтролируемого диффузного потока N2O5 из зоны реакции в этот объем. Все потоки измеряются расходомерами с точностью 0.1 см3 Торр с–1. Отбор пробы в масс-спектрометр проводится в виде молекулярного пучка через отверстие диаметром 0.35 мм в вершине напускного конуса, расположенного соосно с внешней трубкой реактора. Для проведения экспериментов при пониженной температуре реактора были изготовлены две металлические кюветы, заполняемые охлаждающей смесью NaCl/H2O и охватывающие область трубки реактора и компенсирующего объема.
Реагент N2O5 приготавливали заранее в статических условиях по реакции O3 + NO2 → NO3 + O2, NO3 + NO2 → N2O5, затем перемораживали в ампулу криостата при медленной прокачке потоком гелия и хранили при температуре ампулы (178 К). Подача газа-реагента в реактор производится при пропускании постоянного потока Не через ампулу криостата. Концентрацию N2O5 в реакторе изменяли за счет изменения либо температуры криостата, либо потока Не через него. Основным побочным реагентом была двуокись азота, вклад потока которой в поток N2O5 составлял около 5%.
В качестве источника сажи использовали лабораторную горелку, присоединенную к газовой магистрали. Металлический стержень располагали на расстоянии 15–17 см от основания пламени при постоянном вращении стержня вручную. Наибольшая масса навески сажи не превышала предельную величину плотности сажевого покрытия: ρmax ≈ 350 мкг см−2, выше которой необходимо учитывать пористость покрытия [11]. Удельную поверхность сажи определяли экспериментально по методу BET (Brunnauer–Emmett–Teller) [19]. При диффузионном горении метана и в наших условиях нанесения удельная поверхность сажи составляет Sspec = (40 ± 10) м2 · г–1 [20]. Определение Sspec проводилось в тех же условиях, что и при нанесении сажи на металлический стержень.
Стандартизация нанесения сажевого покрытия чрезвычайно важна, поскольку величина удельной поверхности зависит не только от исходного топлива, но и от степени его обогащения и температуры пламени. Так, для одних и тех же топлив в литературе приводятся различные величины удельной поверхности: (15 ± 1) м2 · г–1 для сажи пламени гексана, декана и бензола независимо от вида топлива [21]; (260 ± 40), (175 ± 25) и (120 ± 20) м2 · г–1 для саж обогащенного пламени гексана, толуола и керосина, соответственно [22]; от 20 до 80 м2 · г–1 для сажи пиролиза метана, бензола и толуола в зависимости от температуры пламени и концентрации в потоке газа-носителя [23].
Процедура измерения и обработка данных. Измерение интенсивностей ионных токов проводили в режиме механической модуляции молекулярного пучка и синхронного счета ионов с минимальным временем накопления 5 с. Калибровку масс-спектрометра по NO2 проводили при напуске его измеряемого потока в гелии и измеряемом полном давлении в реакторе. Измерение интенсивности ионного тока NO2 на линии с m/z = 46 проводили при значении энергии ионизирующих электронов Ee = 10.6 эВ. Измерение при этом значении Ee полностью исключает вклад диссоциативной ионизации из N2O5, поскольку эта энергия меньше суммы энергии ионизации NO2 (9.78 эВ) и энергии разрыва связи NO2–NO3 в N2O5 (0.96 эВ).
Абсолютную концентрацию N2O5 в реакторе определяли на основании уравнения
(1)
${\text{[}}{{{\text{N}}}_{{\text{2}}}}{{{\text{O}}}_{{\text{5}}}}{\text{]}} = \frac{{{{w}_{{{\text{He}}}}}}}{{({{w}_{{{\text{He}}}}} + {{w}_{{{\text{He}}ad}}})}}\frac{{{{p}_{{{{{\text{N}}}_{{\text{2}}}}{{{\text{O}}}_{{\text{5}}}}}}}{\text{(}}T{\text{)}}}}{{{{p}_{{\text{a}}}}}}p{{n}_{L}},$При наших скоростях потока и давлении в ректоре кинетика гетерогенного расхода газофазного реагента M на центральном стержне описывается уравнением первого порядка:
(2)
$ - \frac{d}{{d{{t}_{c}}}}{\text{[M(}}{{t}_{c}}{\text{,}}t{\text{)]}} = {{k}_{W}}(t){\text{[}}M({{t}_{c}},t)],$1/kw = 1/$k_{w}^{k}$ + 1/$k_{w}^{d},$ $k_{w}^{k}$ = (γcM/4)(Sef/VR), $k_{w}^{d}$ = 4K(ρ)DМ/$d_{R}^{2}$.
Здесь ρ = dr/dR; K(ρ = 0.15) = 1.7 [25] – безразмерная константа; cM – среднеарифметическая скорость молекул M при данной температуре; DМ – коэффициент диффузии молекул M в гелии; Sef – эффективная площадь покрытия в зоне реакции при выдвижении стержня на длину ΔL; VR – объем реактора, соответствующий этой же длине.
При (dr/dR) $ \ll $ 1 для сажевого покрытия, поверхность которого определяется эффективной площадью, выраженной через массу навески и удельную поверхность, величина $k_{w}^{k}$ определяется уравнением
(3)
$k_{w}^{k} \approx \left( {\gamma {{c}_{M}}{{S}_{{spec}}}{{\rho }_{m}}{\text{/}}{{d}_{R}}} \right)\left( {{{d}_{r}}{\text{/}}{{d}_{R}}} \right),$(4)
$\gamma {\text{(}}t{\text{)}} = \frac{{{\text{ln(}}{{{{I}_{0}}} \mathord{\left/ {\vphantom {{{{I}_{0}}} {I{\text{(}}t{\text{)}}}}} \right. \kern-0em} {I{\text{(}}t{\text{)}}}}{\text{)}}}}{{{{t}_{c}}}}\frac{{d_{R}^{{\text{2}}}}}{{{{c}_{{\text{M}}}}{{S}_{{spec}}}{{\rho }_{m}}{{d}_{r}}}},$Времязависимый захват N2O5на покрытии из метановой сажи. Исследование захвата N2O5 на сажевом покрытии проводилось в диапазоне концентраций [N2O5] = 1.3 ⋅ 1012–3.3 ⋅ 1013 cм–3 при двух температурах: 298 и 255 К, и каждый раз на свежем покрытии. Пример отдельного измерения приведен на рис. 1 при чередовании введений одного и того же сегмента стержня с покрытием в поток газа-реагента при его заданной концентрации. После первого выведения стержня из потока наблюдается десорбция молекул N2O5. При повторном введении этого же участка стержня снова наблюдается расход молекул N2O5, связанный с их повторной адсорбцией. Из-за такой особенности захвата мы были вынуждены каждый раз заменять покрытие на новое при изменении условий проведения эксперимента – концентрации N2O5 или температуры реактора.
Рис. 1.
Зависимость концентрации N2O5 от времени, демонстрирующая захват N2O5 на свежей метановой саже. Условия захвата: [N2O5] = 2.7 · 1012 см–3, Т = 298 К, давление p = 2.8 Торр, ΔL = 10 см, средняя скорость потока гелия u = 74 см · с–1, масса навески сажи на единичную поверхность стержня ρm = 116 мкг · см–2; полые символы – измеряемая концентрация N2O5 при периодическом удалении стержня с покрытием из зоны контакта; сплошные символы – концентрация N2O5 при введенном стержне в поток N2O5.
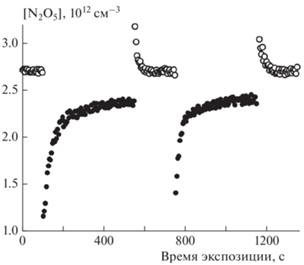
Расчет временнóй зависимости коэффициента захвата проводили по формуле (4) для каждой точки экспериментальной зависимости наиболее интенсивной масс-спектральной линии I46(50 эВ) реагента N2O5. Как отмечено выше, в потоке N2O5 присутствовал побочный продукт, NO2, из-за частичного разложения исходного реагента за время его перемещения из ампулы криостата в реактор. Вклад этого побочного продукта не превышал 5%.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Как установлено в работе [12], начальный захват N2O5 на сажевом покрытии происходит по механизму мономолекулярного распада поверхностного комплекса. Согласно этому механизму коэффициент захвата γ(t) = γ0 exp(–at) должен экспоненциально зависеть от времени экспозиции поверхности к газу-реагенту c постоянной времени 1/a = kθ, равной произведению константы скорости k мономолекулярного распада на долю θ поверхности, занятой адсорбированными молекулами [26]. Как следует из формы временнóй зависимости коэффициента захвата, приведенной на рис. 2, в течение длительного времени экспозиции зависимость оказывается более сложной. Естественно предположить, что захват происходит через несколько последовательных стадий.
Рис. 2.
Временная зависимость коэффициента захвата N2О5, полученная из данных рис. 1: символы – расчет по формуле (4), сплошная кривая – аппроксимация по формуле (8) с параметрами из табл. 1; штриховые кривые – вклады первичного, вторичного и стационарного захватов.
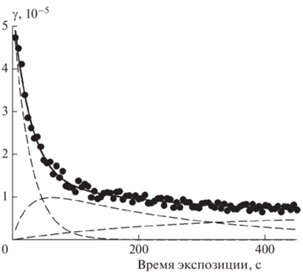
На базе ленгмюровского представления адсорбции ниже приведен простейший механизм реакции, который объясняет временну́ю зависимость коэффициента захвата в течение всего времени экспозиции
(R1)
$\begin{gathered} \hfill {{{\text{N}}}_{{\text{2}}}}{{{\text{O}}}_{{\text{5}}}}\left( {\text{г}} \right) + {{{\text{z}}}_{r}}\left( {{\text{тв}}} \right)\,\,\underset{{{{k}_{d}}}}{\overset{{{{k}_{a}}}}{\longleftrightarrow}}\,\,{{{\text{N}}}_{{\text{2}}}}{{{\text{O}}}_{{\text{5}}}} \ldots {{{\text{z}}}_{r}}\left( {{\text{тв}}} \right), \\ \hfill {{{\text{N}}}_{{\text{2}}}}{{{\text{O}}}_{5}} \ldots {{{\text{z}}}_{r}}\left( {{\text{тв}}} \right)\xrightarrow{{{{k}_{r}}}}{\text{\;}}{{{\text{z}}}_{s}}\left( {{\text{тв}}} \right) + {\text{Продукт }}\left( {\text{г}} \right); \\ \end{gathered} $(R2)
$\begin{gathered} {{{\text{N}}}_{{\text{2}}}}{{{\text{O}}}_{{\text{5}}}}\left( {\text{г}} \right) + {{{\text{z}}}_{s}}\left( {{\text{тв}}} \right)\underset{{{{k}_{d}}}}{\overset{{{{k}_{a}}}}{\longleftrightarrow}}{{{\text{N}}}_{{\text{2}}}}{{{\text{O}}}_{{\text{5}}}} \ldots {{{\text{z}}}_{s}}\left( {{\text{тв}}} \right), \\ {{{\text{N}}}_{{\text{2}}}}{{{\text{O}}}_{5}} \ldots {{{\text{z}}}_{s}}\left( {{\text{тв}}} \right)\,\,\xrightarrow{{{{k}_{s}}}}~\,{{{\text{z}}}_{{fin}}}\left( {{\text{тв}}} \right) + {\text{Продукт }}\left( {\text{г}} \right); \\ \end{gathered} $(R3)
$\begin{gathered} {{{\text{N}}}_{{\text{2}}}}{{{\text{O}}}_{{\text{5}}}}\left( {\text{г}} \right) + {{{\text{z}}}_{{fin}}}\left( {{\text{тв}}} \right)\,\,\underset{{{{k}_{d}}}}{\overset{{{{k}_{a}}}}{\longleftrightarrow}}\,\,{{{\text{N}}}_{{\text{2}}}}{{{\text{O}}}_{5}} \ldots {{{\text{z}}}_{{fin}}}\left( {{\text{тв}}} \right), \\ {{{\text{N}}}_{{\text{2}}}}{{{\text{O}}}_{{\text{5}}}} \ldots {{{\text{z}}}_{{fin}}}\left( {{\text{тв}}} \right)~\xrightarrow{{{{k}_{{fin}}}}}{{{\text{z}}}_{{fin}}}\left( {{\text{тв}}} \right) + {\text{Продукт}}\,\left( {\text{г}} \right). \\ \end{gathered} $Модель предполагает, что захват происходит через три последовательные стадии, идентичные по механизму, но различные по адсорбционно-реакционным характеристикам центров адсорбции. В результате первой, быстрой стадии (R1) из исходного адсорбционного центра zr с поверхностной плотностью [zr] в результате мономолекулярного распада комплекса N2O5…zr с константой скорости kr образуется менее реакционно-способный центр адсорбции zs. В результате второй, медленной стадии (R2) из центров адсорбции zs с константой скорости ks образуются центры zfin, на которых в третьей, заключительной стадии (R3) происходит распад молекул N2O5 без изменения реакционных характеристик этих центров. Состав газофазных продуктов реакций мы не обсуждаем в данной работе.
По этому механизму расход N2O5 в газовой фазе –
(5)
$ - {{V}_{R}}\frac{d}{{dt}}{\text{[}}{{{\text{N}}}_{{\text{2}}}}{{{\text{O}}}_{{\text{5}}}}{\text{]}} = \left[ {{{J}_{{rapid}}}(t) + {{J}_{{slow}}}{\text{(}}t{\text{)}} + {{J}_{{fin}}}{\text{(}}t{\text{)}}} \right]{{S}_{{ef}}}$(6)
$\begin{gathered} {{J}_{{rapid}}}\left( t \right) = {{k}_{r}}\theta \left[ {{{{\text{z}}}_{r}}\left( t \right)} \right],\,\,\,{{J}_{{slow}}}\left( t \right) = {{k}_{s}}\theta \left[ {{{z}_{s}}\left( t \right)} \right], \\ {{J}_{{fin}}}\left( t \right) = {{k}_{{fin}}}\theta \left[ {{{{\text{z}}}_{{fin}}}\left( t \right)} \right]. \\ \end{gathered} $Из сравнения формального уравнения расхода N2O5, выраженного через коэффициент захвата –
Эти реакционные потоки определяются поверхностной плотностью центров адсорбции, зависящей от времени экспозиции:
(7)
$\begin{gathered} - \frac{d}{{dt}}[{{{\text{z}}}_{r}}] = {{k}_{r}}\theta [{{{\text{z}}}_{r}}],\,\,\,\, - \frac{d}{{dt}}[{{{\text{z}}}_{s}}] = {{k}_{s}}\theta [{{{\text{z}}}_{s}}] - {{k}_{r}}\theta [{{{\text{z}}}_{r}}], \\ \frac{d}{{dt}}[{{{\text{z}}}_{{fin}}}] = {{k}_{s}}\theta [{{{\text{z}}}_{s}}], \\ \end{gathered} $Jrapid(t) = krθ[z0]exp(–krθt),
Jslow(t) = ksθ[z0]exp(–ksθt)[1 – exp(–krθt)],
Jfin(t) = kfinθ[z0][1 – exp(–ksθt)],
и явный вид временнóй зависимости коэффициента захвата –
(8)
$\begin{gathered} \gamma \left( t \right) = {{\gamma }_{r}}\exp \left( { - {{a}_{r}}t} \right) + {{\gamma }_{s}}\exp \left( { - {{a}_{s}}t} \right) \times \\ \times \,\,\left[ {1 - \exp \left( { - {{a}_{r}}t} \right)} \right] + {{\gamma }_{{fin}}}\left( {1 - \exp \left( { - {{a}_{s}}t} \right)} \right]. \\ \end{gathered} $Параметры, определяющие эту зависимость, представляют собой комбинацию элементарных констант, описывающих процесс захвата [26]:
(9)
$\begin{gathered} {{\gamma }_{r}} = \gamma _{r}^{{max}}/\left( {1 + {{K}_{L}}\left[ {{{{\text{N}}}_{{\text{2}}}}{{{\text{O}}}_{{\text{5}}}}} \right]} \right), \\ {{\gamma }_{s}} = \gamma _{s}^{{max}}/\left( {1 + {{K}_{L}}\left[ {{{{\text{N}}}_{{\text{2}}}}{{{\text{O}}}_{{\text{5}}}}} \right]} \right), \\ {{\gamma }_{{fin}}} = \gamma _{{fin}}^{{max}}{\text{/}}\left( {1 + {{K}_{L}}\left[ {{{{\text{N}}}_{{\text{2}}}}{{{\text{O}}}_{{\text{5}}}}} \right]} \right); \\ \end{gathered} $(10)
$\begin{gathered} \gamma _{r}^{{max}} = {{\alpha }_{s}}{{k}_{r}}{\text{/}}{{k}_{d}},\,\,\,\gamma _{s}^{{max}} = {{\alpha }_{s}}{{k}_{s}}{\text{/}}{{k}_{d}}, \\ \gamma _{{fin}}^{{max}} = {{\alpha }_{s}}{{k}_{{fin}}}{\text{/}}{{k}_{d}}, \\ \end{gathered} $(12)
$\begin{gathered} \theta = {{K}_{L}}\left[ {{{{\text{N}}}_{{\text{2}}}}{{{\text{O}}}_{{\text{5}}}}} \right]/\left( {1 + {{K}_{L}}\left[ {{{{\text{N}}}_{{\text{2}}}}{{{\text{O}}}_{{\text{5}}}}} \right]} \right), \\ {{K}_{L}} = {{\alpha }_{s}}{{c}_{{{{{\text{N}}}_{{\text{2}}}}{{{\text{O}}}_{{\text{5}}}}}}}{\text{/}}\left( {4{{k}_{d}}\left[ {{{{\text{z}}}_{0}}} \right]} \right), \\ \end{gathered} $Сводные результаты аппроксимации экспериментальных зависимостей γ(t) по формуле (8) при двух температурах реактора и при вариации концентрации N2O5 приведены в табл. 1. На рис. 2 приведен пример такой аппроксимации γ(t) для одной из концентраций N2O5 с соответствующими параметрами из табл. 1. Зависимости этих параметров аппроксимации от концентрации N2O5 при двух температурах представлены на рис. 3–7.
Таблица 1.
Параметры аппроксимации по формуле (8) коэффициента времязависимого захвата N2O5 на свежем сажевом покрытии
| [N2O5], 1012 см–3 | γfin, 10−5 | γr, 10−5 | ar, 10−1 с−1 | γs, 10−5 | as, 10−2 с−1 |
|---|---|---|---|---|---|
| T = 298 K | |||||
| 1.3 | 0.68 ± 0.05 | 5.2 ± 0.8 | 0.18 ± 0.10 | 1.31 ± 0.27 | 0.20 ± 0.10 |
| 1.3 | 0.75 ± 0.10 | 5.7 ± 0.9 | 0.18 ± 0.15 | 1.49 ± 0.20 | 0.10 ± 0.10 |
| 2.7 | 0.57 ± 0.09 | 5.6 ± 0.9 | 0.37 ± 0.15 | 1.40 ± 0.30 | 0.39 ± 0.27 |
| 5.1 | 0.33 ± 0.07 | 5.4 ± 0.8 | 0.46 ± 0.20 | 1.11 ± 0.25 | 0.46 ± 0.13 |
| 8.6 | 0.24 ± 0.06 | 5.3 ± 0.7 | 0.43 ± 0.20 | 1.35 ± 0.20 | 0.64 ± 0.36 |
| 10.4 | 0.30 ± 0.09 | 5.1 ± 0.8 | 0.52 ± 0.21 | 1.20 ± 0.15 | 0.92 ± 0.30 |
| 15.2 | 0.15 ± 0.03 | 4.8 ± 0.7 | 1.00 ±0.20 | 1.25 ± 0.38 | 1.40 ±0.40 |
| 21.5 | 0.14 ±0.03 | 4.8 ±0.7 | 0.96 ± 0.19 | 1.50 ±0.43 | 1.30 ± 0.20 |
| 30.0 | 0.09 ± 0.02 | 4.8 ± 0.5 | 1.20 ± 0.20 | 0.99 ± 0.12 | 1.60 ± 0.40 |
| 33.4 | 0.09 ± 0.02 | 4.5 ± 0.7 | 1.55 ± 0.31 | 1.13 ± 0.39 | 1.90 ± 0.40 |
| T = 255 K | |||||
| 1.3 | 1.05 ± 0.37 | 38 ± 8.0 | 0.53 ± 0.11 | 10.8 ± 3.36 | 1.00 ± 0.20 |
| 3.1 | 1.06 ± 0.37 | 27 ± 6.0 | 0.70 ± 0.14 | 12.2 ± 4.73 | 1.80 ± 0.30 |
| 6.3 | 0.51 ± 0.09 | 21 ± 4.0 | 1.08 ± 0.22 | 6.30 ± 1.20 | 2.00 ± 0.30 |
| 10.0 | 0.41 ± 0.08 | 17 ± 3.0 | 1.37 ± 0.27 | 6.24 ± 3.73 | 1.85 ± 0.40 |
| 14.0 | 0.23 ± 0.03 | 12 ± 3.0 | 1.37 ± 0.27 | 3.90 ± 1.90 | 2.64 ± 0.50 |
| 17.2 | 0.22 ± 0.04 | 12 ± 3.0 | 1.36 ± 0.27 | 3.31 ± 0.67 | 2.53 ± 0.40 |
| 23.6 | 0.16 ± 0.03 | 9.0 ± 3.0 | 1.42 ± 0.28 | 3.13 ± 0.54 | 2.20 ±0.20 |
| 26.7 | 0.15 ±0.03 | 6.6 ± 1.6 | 1.65 ± 0.33 | 2.16 ± 0.60 | 2.26 ± 0.60 |
Рис. 3.
Зависимость параметра γr времязависимого захвата N2O5 на свежей метановой саже от [N2O5]: символы – данные из табл. 1 при 298 (полые) и 255 К (сплошные); сплошные линии – аппроксимация по формуле (9) с параметрами $\gamma _{r}^{{max}}$ и KL,r из табл. 2.
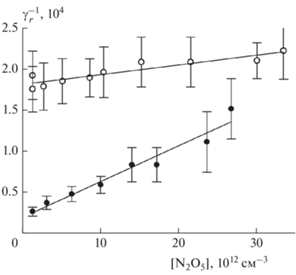
Рис. 4.
Зависимость параметра γs времязависимого захвата N2O5 на свежей метановой саже от [N2O5]: символы – данные из табл. 1 при 298 (полые) и 255 К (сплошные); сплошные линии – аппроксимация по формуле (9) с параметрами $\gamma _{s}^{{max}}$ и KL,s из табл. 2.
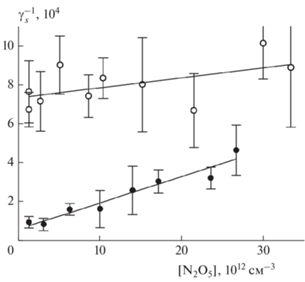
Рис. 5.
Зависимость параметра γfin стационарного захвата N2O5 на свежей метановой саже от [N2O5]: символы – данные из табл. 1 при 298 (полые) и 255 К (сплошные); сплошные линии – аппроксимация по формуле (9) с параметрами $\gamma _{{fin}}^{{max}}$ и KL,fin из табл. (2).
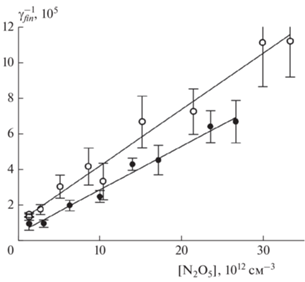
Рис. 6.
Зависимость параметра ar времязависимого захвата N2O5 на свежей метановой саже от [N2O5]: символы – данные из табл. 1 при 298 (полые) и 255 К (сплошные); сплошные кривые – аппроксимация по формуле (11) с параметрами kr и KL,r из табл. 2.
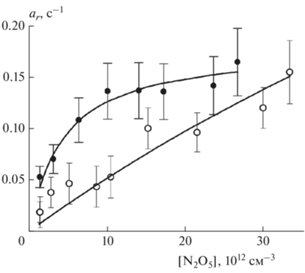
Рис. 7.
Зависимость параметра as времязависимого захвата N2O5 на свежей метановой саже от [N2O5]: символы – данные из табл. 1 при 298 (полые) и 255 К (сплошные); сплошные кривые – аппроксимация по формуле (11) с параметрами ks и KL,s из табл. 2.
Зависимости компонентов γr, γs и γfin коэффициента захвата из формулы (8) от концентрации N2O5 показаны на рис. 3–5. Они соответствуют заявленному механизму мономолекулярного распада поверхностного комплекса – коэффициент захвата уменьшается при увеличении концентрации и стремится к предельному значению при [N2O5] → 0. Из аппроксимации этих зависимостей по формуле (9) для каждой из них мы находим два параметра: максимальное значение γmax коэффициента захвата и соответствующий коэффициент Ленгмюра KL, который определяет изотерму адсорбции для данного типа поверхностных центров. Сводные данные по этим параметрам аппроксимации приведены в табл. 2.
Таблица 2.
Механизм многостадийного захвата N2O5 на свежем сажевом покрытии. Параметры аппроксимации по формулам (9) и (11) коэффициентов времязависимого захвата
| T, K | Первая, быстрая стадия | Вторая, медленная стадия | Стационарный захват | ||||||
|---|---|---|---|---|---|---|---|---|---|
| $\gamma _{r}^{{max}}$, 10−4 | KL,r, 10−15 см3 | kr, с−1 | $\gamma _{s}^{{max}}$, 10−4 | KL,s, 10−15 см3 | ks, 10–2 с−1 | $\gamma _{{fin}}^{{max}}$, 10−5 | KL,fin, 10−13 см3 | kfin, с−1 | |
| 255 | 5.3 ± 1.8 | 235 ± 84 | 0.18 ± 0.04 | 1.8 ± 0.7 | 255 ± 100 | 3 ± 0.2 | 2.4 ± 1.1 | 5.7 ± 2.8 | − |
| 298 | 0.55 ± 0.1 | 6.6 ± 1.5 | 0.84 ± 0.05 | 0.14 ± 0.01 | 7.0 ± 3.8 | 10.5 ± 0.6 | 1.0 ± 0.3 | 3.1 ± 1.0 | − |
Зависимости от концентрации N2O5 параметров ar и as коэффициента захвата, характеризующих их временну́ю зависимость, приведены на рис. 6 и 7. Из аппроксимации этих зависимостей по формуле (11) определены константы скорости соответствующих мономолекулярных распадов. Значения этих параметров также приведены в табл. 2. Отметим, что эти искомые константы были единственными подгоночными параметрами, поскольку коэффициенты KL, входящие в сомножители θ из формулы (12), взяты из результатов аппроксимации γr и γs.
В предположении аррениусовской зависимости констант скорости десорбции, kd = νd exp(–Qad/RT), и мономолекулярного распада, kr = Ar exp(–Ea/RT), из отношения KL(255 K)/KL(298 K) для всех трех стадий захвата с учетом зависимости (12) KL от kd находим теплоту адсорбции на центрах zr, zs и zfin. Из отношения γmax(255 K)/γmax(298 K) c учетом зависимости (10) γmax от kd и kr находим разность Qad – Ea. Результаты оценки приведены в табл. 3. В последнем столбце табл. 3 приведены энергии активации этих же реакций. Они получены независимо из отношений kr(298 K)/kr(255 K) и ks(298 K)/ ks(255 K) и величин этих констант их табл. 2. Как следует из сравнения Ea, полученных независимо двумя разными способами, они совпадают в пределах ошибки вычисления.
Таблица 3.
Механизм многостадийного захвата. Теплоты адсорбции N2O5 на сажевом покрытии и энергии активации констант скорости
| Стадия захвата | Qad, кДж · моль–1 | Qad − Ea, кДж · моль–1 | Ea, кДж · моль–1 |
|---|---|---|---|
| Быстрая | 54 ± 6 | 33.3 ± 5.7 | 22.6 ± 3.6 |
| Медленная | 54 ± 10 | 38 ± 6 | 18.4 ± 1.6 |
| Стационарная | 10 ± 8 | 12.7 ± 8 | − |
Теплоты адсорбции молекул N2O5 на исходных, zr, и модифицированных, zs, центрах идентичны. Энергии активации констант скорости мономолекулярного распада поверхностных комплексов на этих центрах также совпадают в пределах ошибки вычисления. Таким образом, наблюдаемое различие скоростей захвата на первой и второй стадиях обусловлено разницей предэкспонентов этих констант скорости в 40 раз. Количественные данные табл. 3 по стационарной стадии захвата носят оценочный характер, поскольку адсорбционные характеристики сильно отличаются от аналогичных характеристик для предыдущих стадий и противоречат условию вывода формулы (8) для временнóй зависимости коэффициента захвата.
ЗАКЛЮЧЕНИЕ
Введение в механизм захвата N2O5 медленной стадии позволяет количественно описать этот процесс в течение всего времени экспозиции. В результате модельного описания захвата N2O5 на свежем сажевом покрытии получена совокупность элементарных параметров, позволяющих моделировать этот захват в условиях тропосферы при произвольных концентрациях N2O5 и температуре. В качестве иллюстрации сравним процесс захвата при Т = 298 К и двух предельных концентрациях N2O5: 50 ppt и 3.8 ppb, на сажевых частицах аэрозоля коагуляционной моды с характерным размером последних 0.1–1 мкм. Время жизни этих частиц аэрозоля определяется скоростью их осаждения и в нижней тропосфере составляет порядка 8 сут [28]. При минимальной концентрации 50 ppt усредненное значение кинетического предела коэффициента захвата по времени жизни аэрозоля, вычисленное по формуле (8) и данным табл. 2, составляет 2.2 · 10–5. Этот же параметр при концентрации 3.8 ppb оказывается в 2 раза меньше и на протяжении всего времени жизни аэрозоля будет определяться практически стационарным захватом с коэффициентом захвата γfin. Начальная величина коэффициента захвата для обеих предельных концентраций остается неизменной и равной 5.5 · 10–5 в расчете на истинную ВЕТ-поверхность. В реальных условиях многокомпонентного состава малых газовых составляющих тропосферы это означает, что и реакционные характеристики поверхностных центров по отношению к захвату других газов-реагентов будут также времязависимыми.
Эта работа выполнена в рамках госзадания FFZE-2022-0008 (регистрационный номер 1021051302551-2-1.3.1;1.4.7;1.6.19).
Список литературы
Chang W.L., Bhave P.V., Brown S.S. et al. // Aerosol Sci. Technol. 2011. V. 45. P. 665; https://doi.org/10.1080/02786826.2010.551672
Wagner N.L., Riedel T.P., Young C.J. et al. // J. Geophys. Res. 2013. V. 118D. P. 9331; https://doi.org/10.1002/jgrd.50653
Wang R., Tao S., Shen H. et al. // Environ. Sci. Technol. 2014. V. 48. P. 6780; https://doi.org/10.1021/es5021422
Berner A., Sidla S., Galambos Z. et al. // J. Geophys. Res. Atmospheres. 1996. V. 101. P. 19559; https://doi.org/10.1029/95JD03425
Pohl K., Cantwell M., Herckes P., Lohmann R. // Atmos. Chem. Phys. 2014. V. 14. P. 7431; https://doi.org/10.5194/acp-14-7431-2014,2014
Sander S.P., Abbatt J., Barker R. et al. JPL Publication 10-6. “Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies” No 17, 2011; http://jpldataeval.jpl.nasa.gov
Riedel T.P., Bertram T.H., Ryder O.S. et al. // Atmos. Chem. Phys. 2012. V. 12. P. 2959; https://doi.org/10.5194/acp-12-2959-2012,2012
Brower L., Rossi M.J., Golden D.M. // J. Phys. Chem. 1986. V. 90. P. 4599; https://doi.org/10.1021/j10041a025
Longfellow C.A., Ravishankara A.R., Hanson D.R. // J. Geophys. Res. Atmospheres. 2000. V. 105. P. 24345; https://doi.org/10.1029/2000JD900297
Saathoff H., Naumann K.-H., Riemer N. et al. // Geophys. Res. Lett. 2001. V. 28. P. 1957; https://doi.org/10.1029/2000GL012619
Karagulian F., Rossi M.J. // J. Phys. Chem. A. 2007. V. 111. P. 1914; https://doi.org/10.1021/jp0670891
Зеленов В.В., Апарина Е.В., Каштанов С.А., Шардакова Э.В. // Хим. физика. 2016. Т. 35. № 4. С. 78; https://doi.org/10.7868/S0207401X16040129
Ammann M., Pöschl U., Rudich Y. // Phys. Chem. Chem. Phys. 2003. V. 5. P. 351; https://doi.org/10.1039/b208708a
Ammann M., Pöschl U. // Atmos. Chem. Phys. 2007. V. 7. P. 6025; www.atmos-chem-phys.net/7/6025/2007/
Pöschl U., Rudich Y., Ammann M. // Atmos. Chem. Phys. 2007. V. 7. P. 5989; www.atmos-chem-phys.net/ 7/5989/2007/
Berkemeier T., Ammann M., Krieger U.K. et al. // Atmos. Chem. Phys. 2017. V. 17. P. 8021; https://doi.org/10.5194/acp-17-8021-2017
Травин С.О., Скурлатов Ю.И., Рощин А.В. // Хим. физика. 2020. Т. 39. № 2. С. 3; https://doi.org/10.31857/S0207401X20020144
Зеленов В.В., Апарина Е.В. // Хим. физ. 2021. Т. 40. № 5. С. 55; https://doi.org/10/31857/S0207401X21050149
Травин С.О., Громов О.Б., Утробин Д.В., Рощин А.В. // Хим. физика. 2019. Т. 38. № 11. С. 5; https://doi.org/10.1134/S0207401X19110116
Зеленов В.В., Апарина Е.В., Каштанов С.А., Шардакова Э.В. // Хим. физика. 2015. Т. 34. № 3. С. 87; https://doi.org/10.7868/S0207401X15030140
Aubin D.G., Abbatt J.P.D. //J. Phys. Chem. 2007. V. 111. P. 6263; https://doi.org/10.1021/jp068884h
Lelievre S., Bedjanian Yu., Laverdet G., Le Bras G. // J. Phys. Chem. A. 2004. V. 108. P. 10807; https://doi.org/10.1021/jp0469970
Tesner P.A., Shurupov S.V. // Combust. Sci. Technol. 1995. V. 105. P. 147; https://doi.org/10.1080/00102209508907744
Ефимов А.И., Белорукова Л.П., Василькова И.В., Чечев В.П. Свойства неорганических соединений. Справочник. Л.: Химия, 1983.
Gershenzon Yu.M., Grigorieva V.M., Ivanov A.V., Remorov R.G. // Faraday Discuss. 1995. V. 100. P. 83.
Laidler K.J. // Chemical kinetics. 2nd ed. N.Y.: McGraw-Hill, 1965.
Berkemeier T., Huisman A.J., Ammann M. et al. // Atmos. Chem. Phys. 2013. V. 13. P. 6663; https://doi.org/10.5194/acp-13-6663-2013
Bond T.C., Streets D.G., Yarber K.F. et al. // J. Geophys. Res. 2004. V. 109. D14203; https://doi.org/10.1029/2003JD003697
Дополнительные материалы отсутствуют.
Инструменты
Химическая физика