

Журнал высшей нервной деятельности им. И.П. Павлова, 2022, T. 72, № 3, стр. 293-316
Спектр синдромальных нарушений при экспансии CGG-повторов промотора гена FMR1: патогенетические механизмы и клинические проявления
Д. С. Переверзева 1, С. А. Тюшкевич 1, Е. В. Улас 2, Н. Л. Горбачевская 1, 3, *
1 Московский государственный психолого-педагогический университет (ФГБОУ ВО МГППУ)
Москва, Россия
2 Институт белка РАН
Пущино, Россия
3 Научный центр психического здоровья (ФГБНУ НЦПЗ)
Москва, Россия
* E-mail: gorbachevskaya@yandex.ru
Поступила в редакцию 21.11.2020
После доработки 01.03.2021
Принята к публикации 02.03.2021
- EDN: HHQDZG
- DOI: 10.31857/S004446772203008X
Аннотация
Спектр заболеваний, ассоциированных с ломкой хромосомой Х (FXSD) объединяет следующие клинические синдромы: синдром умственной отсталости, сцепленной с ломкой хромосомой Х (fragile X syndrome, FXS), синдром тремора и атаксии (FXTAS), синдром первичной овариальной недостаточности (FXPOI), группа нейропсихиатрических нарушений (FXAND). Причиной их возникновения являются динамические мутации гена FMR1, вызванные экспансией тринуклеотидных повторов в промоторе гена. Синдром FXS (умственная отсталость, РАС, СДВГ) возникает при полной мутации гена FMR1 (увеличение количества CGG-повторов свыше 200), сопровождающейся полным или частичным подавлением экспрессии белка FMRP. Клинические проявления FXTAS, FXPOI, FXAND могут возникнуть у лиц-носителей премутации FMR1 (диапазон значений количества CGG-повторов от 55 до 200). К основным патогенетическим механизмам этих заболеваний относят снижение экспрессии FMRP и накопление мРНК FMR1, содержащей увеличенное количество повторов. Настоящий обзор посвящен анализу гено-фенотипических отношений внутри спектра состояний, ассоциированных с динамическими мутациями FMR1. В статье представлен анализ взаимосвязи молекулярных механизмов (дефицит FMRP, трансляция атипичных изоформ, усиление функции мРНК FMR1) и клинических проявлений (уровень когнитивного развития, тяжесть РАС, тяжесть симптомов FXTAS, FXPOI и FXAND).
ВВЕДЕНИЕ
К синдромам, вызываемым динамическими мутациями гена FMR1, относится синдром умственной отсталости, сцепленной с ломкой хромосомой Х (Fragile X syndrome, FXS), синдром тремора и атаксии (fragile X-associated tremor/ataxia syndrome, FXTAS), синдром первичной овариальной недостаточности (fragile X-associated premature ovarian insufficiency, FXPOI), группа нейропсихиатрических нарушений (fragile X- associated neuropsychiatric disorders, FXAND). Мутации FMR1 могут также стать причиной синдрома дефицита внимания и гиперактивности (СДВГ) в детском возрасте, эндокринных и аутоиммунных нарушений (для обзора см. (Переверзева и др., 2019)).
Ген FMR1 кодирует белок FMRP, играющий важную роль в созревании нервных клеток. Наиболее частые мутации этого гена связаны с увеличением числа копий тринуклеотидных CGG-повторов в промоторе. Данные мутации гена приводят к полному или частичному подавлению экспрессии белка FMRP. В наиболее общем виде выделяют три типа нарушений функционирования гена FMR1, обусловленных накоплением в нем CGG-повторов: “серая зона” (число повторов от 40 до 54), премутация (число повторов от 55 до 199 включительно) и полная мутация (число повторов 200 и более). Полная мутация гена вызывает ингибирование экспрессии FMRP и, как следствие, развитие синдрома FX. Это тяжелое заболевание, центральное место в клинической картине которого занимают нарушения психического развития, интеллектуальная недостаточность и расстройства аутистического спектра (РАС). Премутация гена FMR1 приводит к умеренному дефициту белка FMRP и избытку транскрипта гена FMR1, накопление которого имеет нейротоксический эффект. Премутация ассоциирована с такими синдромами, как FXTAS, FXPOI, FXAND, эндокринными заболеваниями и аутоиммунными расстройствами, а также нарушениями развития в детском возрасте: расстройствами аутистического спектра (РАС), синдромом дефицита внимания и гиперактивности (СДВГ), трудностями школьного обучения (ТШО).
Клинические проявления при состояниях, ассоциированных с мутациями гена FMR1, несмотря на свое разнообразие, имеют ряд пересечений. Степень дефицита FMRP различается в зависимости от типа мутации и пола индивидуума и значимо коррелирует с уровнем когнитивного развития (Kim et al., 2019; Loesch et al., 2004). Точно так же существует зависимость между количеством повторов в промоторе FMR1, количеством транскрипта гена и тяжестью синдромов FXTAS, FXPOI, FXAND (Loesch, Hagerman, 2012).
Настоящая статья посвящена описанию гено-фенотипических взаимосвязей между количеством тринуклеотидных повторов в промоторе гена FMR1 и особенностями клинической картины при FXSD (Fragile X spectrum disorders, спектр заболеваний, ассоциированных с ломкой хромосомой Х). Особое внимание мы уделяем патогенетическим механизмам, лежащим в основе формирования фенотипических проявлений данных заболеваний. Представлен анализ данных о гетерогенности клинических проявлений, рисках и тяжести развития когнитивного дефекта, РАС, СДВГ, FXTAS, FXPOI, FXAND, в зависимости от таких генетических особенностей, как тип мутации в гене FMR1, гомо- либо гетерозиготное состояние мутации, наличие либо отсутствие генетического мозаицизма.
1. СИНДРОМЫ, АССОЦИИРОВАННЫЕ С МУТАЦИЯМИ ГЕНА FMR1
1.1. Синдром FX, ассоциированный с полной мутацией гена FMR1
Синдром умственной отсталости, связанный с ломкой хромосомой Х (fragile X syndrome, FXS – синдром FX, синдром Мартина–Белл), – это тяжелое заболевание, характеризующееся выраженным снижением интеллектуального развития, расстройствами аутистического спектра, задержкой и своеобразием развития речи (для обзора см. Salcedo-Arellano et al., 2020). Распространенность синдрома составляет 1 : 7000 среди мужчин и 1 : 11 000 среди женщин. 98–100% мальчиков с синдромом FX имеют умственную отсталость (Тюшкевич и др., 2010; Budimirovic, 2016), причем в половине случаев IQ не превышает 50 ед. (Hessl et al., 2009). Исключение составляют случаи мозаицизма при FXS, для которых характерны более мягкие проявления интеллектуальной недостаточности (IQ 70 и выше). Картина когнитивных нарушений у женщин в большей степени вариативна. Средний показатель общего интеллекта составляет 74 балла (de Vries, 1996). Около 15% имеют средние нормативные показатели интеллекта (выше 85 баллов) (Sterling, Abbeduto, 2012). Это связано с тем, что женщины в подавляющем большинстве случаев гетерозиготны по мутации в FMR1, поскольку имеют в своем геноме две копии хромосомы X (на которой локализован ген FMR1), т.е. наряду с мутантным аллелем у них присутствует и аллель дикого типа. В силу процесса X-инактивации у женщин мутантная копия гена будет представлена в одной части клеток, в то время как в другой части экспрессируется копия гена дикого типа. Таким образом, тяжесть болезни у женщины будет зависеть от соотношения в организме этих двух типов клеток (Kirchgessner et al., 1995; Heine-Suñer et al., 2003). Гиперактивность и нарушение внимания (у 80% мальчиков и до 40% девочек), а также социальная тревожность (до 86%) являются преобладающей симптоматикой в картине поведенческих нарушений при FXS. Расстройства в спектре аутизма диагностируются в половине случаев у детей с FXS, при этом степень и глубина их проявлений сильно варьирует от случая к случаю. В дополнение к интеллектуальной недостаточности и поведенческим расстройствам при синдроме FX с высокой частотой отмечаются фенотипические проявления, такие как удлиненное лицо, широкий лоб, оттопыренные уши, макроцефалия, гипермобильность суставов и макроорхидизм (в 80% случаев встречается как минимум один из перечисленных признаков). К неврологическим особенностям синдрома относят эпи-активность, встречающуюся примерно в 15–20% случаев в детском возрасте (Rajaratnam et al., 2017).
1.2. Синдромы, ассоциированные с премутацией гена FMR1
Распространенность премутации гена FMR1 в популяции крайне велика и составляет по различным оценкам от 1 : 110 до 1 : 270 среди лиц женского пола, от 1 : 250 до 1 : 830 среди лиц мужского пола (Hagerman et al., 2017). С премутацией ассоциированы синдром тремора и атаксии (FXTAS), синдром первичной овариальной недостаточности (FXPOI), нейропсихиатрические нарушения (FXAND), а также нарушения развития в детском возрасте.
1.2.1. Синдром тремора и атаксии (FXTAS – fragile X-associated tremor/ataxia syndrome)
В старшем возрасте (старше 50 лет) у мужчин (в среднем по группе до 40% встречаемости) и у женщин (не более 20%) развивается синдром тремора и атаксии, характеризующийся прогрессирующим тремором при произвольных движениях, нарушениями походки, паркинсонизмом, периферической невропатией, апноэ, мигренями (Rodriguez-Revenga et al., 2009). В ряде исследований была показана возрастнозависимая пенетрантность FXTAS, характерная как для мужчин, так и для женщин с премутацией: в возрасте 50–59 лет FXTAS проявляется у 8–17% носителей премутации, у 38% – в возрасте 60–69 лет, у 47% – в возрасте 70–79 лет, у 75% – после 80 лет (описаны единичные случаи в данном возрастном периоде) (Jacquemont et al., 2004; Rodriguez-Revenga et al., 2009). Однако стоит заметить, что приведенная возрастная динамика проявления FXTAS наиболее полно описывает мужскую группу с премутацией, тогда как пенетрантность FXTAS у женщин обычно ниже, при этом в результате X-инактивации тяжесть клинических симптомов у женщин прямо пропорциональна доле в их организме клеток с активной хромосомой X, несущей ген FMR1 с премутацией. У лиц – носителей премутации, которые не имеют выраженных симптомов нейродегенеративных нарушений, также отмечается снижение с возрастом показателей исполнительных когнитивных функций вплоть до развития деменции у лиц с FXTAS (Cornish et al., 2009; Hashimoto et al., 2010). У мужчин в первую очередь страдают функции планирования, тормозный контроль и рабочая память. У женщин отмечается дефицит зрительно-пространственных представлений, конструктивных способностей (Lozano et al., 2014).
1.2.2. Синдром первичной овариальной недостаточности у женщин (FXPOI – fragile X-associated primary ovarian insufficiency)
FXPOI наблюдается примерно у 20% женщин с премутацией и составляет около 2% от всех случаев овариальной недостаточности среди женщин. Основными симптомами являются нерегулярный менструальный цикл, наступление ранней менопаузы, бесплодие, повышенный уровень фолликулостимулирующего гормона (ФСГ) (Welt et al., 2004; Nelson, 2005).
1.2.3. Нейропсихиатрические нарушения (синдром FXAND – Fragile X-Associated Neurodevelopmental Disorders)
Самыми частыми клиническими проявлениями, ассоциированными с премутацией гена FMR1, являются психоэмоциональные проблемы, которые были объединены Р. Хагерман в отдельный синдром, получивший название FXAND.
Было показано, что у женщин с премутацией существенно повышен риск развития тревожного расстройства (до 70%), депрессии и других нарушений настроения (43% для лиц с премутацией, 31% для контрольной группы), хронической боли и фибромиалгии (26–46% для лиц с премутацией), панических атак (8.6% – лица с премутацией, 3.2% – контрольная группа), социофобии (Hagerman et al., 2018). Помимо этого отмечается высокий риск формирования зависимостей (в т.ч. алкогольной и наркотической), синдрома хронической усталости.
Хотя аутоиммунные болезни (АиБ) не являются нейропсихиатрическими проблемами, они также связаны с премутацией гена FMR1. Интересно, что АиБ возникают преимущественно у женщин-носительниц, тогда как у мужчин с премутацией аутоиммунные проблемы встречаются редко. Среди лиц с премутацией наиболее распространено аутоиммунное заболевание щитовидной железы (24.4%), за которым следует фибромиалгия (10.2%) и синдром раздраженного кишечника (9.9%). В своей работе Ялнапуркар с соавторами предположили, что АиБ могут усугубить эмоциональные нарушения и ускорить наступление FXTAS (Jalnapurkar et al., 2015). Характерно, что депрессия часто предшествует синдрому FXTAS и может рассматриваться как продромальный признак.
1.2.4. Нарушения развития, проявляющиеся в детском возрасте
Число исследований, посвященных специфике развития в детском возрасте у носителей премутации, крайне ограниченно. Обобщенные данные указывают на незначительную в сравнении с нормой задержку в общем развитии в раннем возрасте, на высокий риск развития РАС и СДВГ (Bailey et al., 2008, Clifford et al., 2007; Hagerman, Hagerman, 2004). Так, в четырехлетнем лонгитюдном исследовании (первый измерительный срез был проведен в трехмесячном возрасте детей) было показано, что профили когнитивного, адаптивного, коммуникативного и социального развития у детей с премутацией статистически не отличались от нормы. Однако для детей с премутацией был характерен своеобразный профиль сенсорного восприятия: с одной стороны, отмечались преобладание сенсорного поиска в поведении и гипореактивность на сенсорные раздражители разных модальностей, с другой стороны, было обнаружено увеличение гиперчувствительности с возрастом, что не характерно для нормотипичого развития (Wheeler et al., 2016; Raspa, 2018). Выявленные особенности сенсорного восприятия, по мнению авторов исследования, могут быть рассмотрены как ранние маркеры регуляторных и эмоционально-личностных проблем (СДВГ, РАС, тревожность) в более позднем возрасте. В другом сравнительном исследовании зрительных предпочтений в разных группах детей (типичное развитие, синдром Дауна, синдром FX, премутация, средний возраст испытуемых составил 17.5 мес) было показано, что у детей с премутацией отмечаются более высокие пороги зрительного опознания (уровень контрастной чувствительности 42% и выше) движущихся стимулов второго порядка (определяемых текстурой), чем в контрольной группе. Схожие результаты были получены в группе детей с полной мутацией и синдромом Дауна, что, как полагают авторы работы, указывает на существование некоторого общего дефицита в развитии пространственно-временных представлений при различных вариантах нарушенного развития (Gallego et al., 2014). Еще одним высокочастотным проявлением в детской группе у носителей премутации являются симптомы РАС. Так, аутистические проявления встречаются, по разным данным, у 14–19% мальчиков и 1–5% девочек, что существенно превышает данные по популяции в целом. Гиперактивность и нарушение внимания присутствуют примерно в 40% случаев (Farzin et al., 2006).
1.3. “Серая зона”
Так называемая “серая зона” – состояние, связанное с количеством CGG-повторов в FMR1 в диапазоне 41–54, – также оказывается ассоциирована с рядом клинических особенностей. Несмотря на то, что фенотип индивидуумов с таким количеством повторов не отличается от нормы, существуют исследования, которые показывают повышенный риск развития паркинсонизма как у мужчин, так и у женщин (Loesch et al., 2018; Hall et al., 2020), а также невынашиваемости беременности (Dean et al., 2019). Согласно данным скрининга новорожденных в США частота встречаемости “серой зоны” составляет 1 : 66 среди лиц женского пола и 1 : 112 среди лиц мужского пола (Tassone et al., 2012).
2. ПАТОГЕНЕТИЧЕСКИЕ МЕХАНИЗМЫ, СВЯЗАННЫЕ С ДИНАМИЧЕСКИМИ МУТАЦИЯМИ В ГЕНЕ FMR1
2.1. Ген FMR1: мутации
Ген FMR1 имеет высоко консервативную последовательность. У человека он локализован в длинном плече хромосомы X, в участке Xq27.3, и имеет следующие геномные координаты (GRCh38): X:147,911,918-147,951,124. FMR1 на высоком уровне экспрессируется в эмбрионах человека. мРНК FMR1 синтезируется в мозге плода на ранних стадиях эмбриогенеза в пролиферирующих и мигрирующих клетках нервной системы и сетчатки, а также в хрящевых структурах и печени. В мозге 25‑недельного эмбриона человека мРНК FMR1 продуцируется в наибольших количествах в холинергических нейронах базального ядра и пирамидных нейронах гиппокампа (Abitbol et al., 1993). Постнатально на наиболее высоком уровне мРНК FMR1 синтезируется в мозге, половых железах, эзофагеальном эпителии, вилочковой железе, глазах и селезенке (Hinds et al., 1993). Ген включает в себя 17 экзонов. Результатом альтернативного сплайсинга является образование нескольких вариантов мРНК FMR1, транслируемых в различные изоформы FMRP, каждая из которых имеет собственные биохимические свойства (Ashley et al., 1993).
Транскрипция FMR1 находится под контролем промотора данного гена – последовательности ДНК, в окружении которой локализован сайт начала транскрипции FMR1. Промотор FMR1 представляет собой небольшой участок последовательности ДНК, располагающийся от 355-го нуклеотида перед областью экспансии CGG-повторов (5') до 60-го нуклеотида после данной области (3') (Kumari et al., 2001). Таким образом, промотор гена включает в себя область экспансии тринуклеотидных повторов, а также предшествующий ей богатый CpG-динуклеотидами островок, внутри которого находится сайт старта транскрипции FMR1. При этом в промоторе отсутствует канонический TATA-бокс (Drouin et al., 1997). CpG-островок FMR1 содержит 52 CpG динуклеотида, которые в случае FXS подвергаются метилированию, что приводит к “молчанию” гена FMR1 (Pietrobono et al., 2002). При FXS подавление экспрессии FMR1 происходит на 11-й неделе эмбриогенеза: в этот момент в случае наличия 200 или более CGG-повторов в промоторе FMR1, как правило, запускаются эпигенетические механизмы метилирования ДНК (присоединения метильных групп к цитозинам в составе CpG-островка) и внесения специфических модификаций в гистоновые белки (Colak et al., 2014). Впрочем, как недавно было показано, изменения паттернов метилирования, связанные с накоплением 200 и более CGG-повторов в FMR1, затрагивают не только область CpG-островка промотора FMR1. Точная граница метилирования ДНК была обнаружена в 5' области гена, предшествующей сайту начала транскрипции FMR1, протяженностью от 650-го до 800-го нуклеотида перед участком CGG-повторов FMR1 (Naumann et al., 2009), – у типично развивающихся индивидуумов эта граница отделяет метилированную ДНК, расположенную перед промоторной областью FMR1, от неметилированного промотора FMR1. Данный участок получил название FREE1 (fragile X-related element 1) (Godler et al., 2010). У индивидуумов с 200 или более CGG-повторами в FMR1 эта граница в норме не подверженной метилированию ДНК в большинстве случаев утрачивается: метилирование распространяется на участок FREE1, а также в CpG-островок – область, где располагается участок старта транскрипции FMR1. Метилирование FREE1 коррелирует с дефицитом FMRP в крови индивидуумов с FXS (Godler et al., 2011).
Схожая граница между ДНК, подверженной метилированию и не затронутой эпигенетическими модификациями транскрипционного сайленсинга, присутствует также в участке, расположенном внутри интрона 1 гена FMR1, и данная граница также утрачивается у людей с FXS, позволяя метилированию распространяться на весь интрон 1 гена FMR1 и далее от 3'- к 5'- последовательности ДНК, в так называемую область FREE2 (fragile X-related element 2) (Godler, 2012; Godler et al., 2010). Эта область включает в себя 3'-концевой фрагмент экзона 1 и 5'-концевой фрагмент интрона 1 кодирующей последовательности гена FMR1. Метилирование в FREE1, FREE2 и области CpG-островка FMR1 при наличии 200 или более CGG-повторов происходит согласованно, что было показано для различных типов тканей, и приводит к подавлению экспрессии FMRP (Godler et al., 2010).
В последние годы активно изучается роль метилирования интрона 1 гена FMR1 как прогностического критерия тяжести заболевания у индивидуумов с полной мутацией и премутацией в данном гене (Godler et al., 2012, Cornish et al., 2015; Shelton et al., 2017).
В норме число CGG в гене FMR1 составляет от 5 до 44 и сопровождается наличием AGG-триплетов после каждых 9–10 CGG-повторов (в общей популяции почти в 95% аллелей FMR1 область CGG-повторов имеет одно или два прерывания AGG). Считают, что наличие AGG триплетов придает стабильность числу CGG-повторов в процессе гаметогенеза и, соответственно, при передаче от родителя потомству, предотвращая формирование вторичных структур ДНК (таких как шпильки) при ее репликации. Потеря вставок AGG делает повтор CGG способным к экспансии (Юдкин и др., 2015).
В зависимости от числа CGG-повторов заболевания, связанные с экспансией тринуклеотидных повторов в гене FMR1, имеют следующие варианты. Аллели, несущие от 45 до 54 повторов CGG, называются промежуточными. Они располагаются в диапазоне так называемой “серой зоны”. Эти аллели широко распространены в популяции (0.8–3.6% с некоторой географической вариабельностью, ~ 1 : 66 среди женщин, ~ 1 : 112 среди мужчин) и имеют высокий риск увеличения числа тринуклеотидных повторов при передаче потомству, если количество CGG-триплетов более пятидесяти и при этом число не прерывающихся вставками AGG CGG-триплетов превышает 35 (Villate et al., 2020).
Увеличение количества повторов CGG свыше 55 связывают с двумя видами нарушений функционирования гена FMR1: премутация (количество повторов от 55 до 199 включительно (~1 : 200 среди женщин; ~1 : 800 среди мужчин), полная мутация (количество повторов 200 и более). Как уже было описано выше, полная мутация гена в большинстве случаев приводит к метилированию участков FREE1, FREE2 и области CpG-островка промотора FMR1, что, в свою очередь, влечет за собой ингибирование трансляции белка FMRP.
2.2. Накопление и дефицит транскриптов FMR1
Механизмы патогенеза при премутации в FMR1 отличаются от механизмов, лежащих в основе развития FXS. В случаях премутации в FMR1 наиболее важными патогенетическими факторами являются: опосредованная повышенной экспрессией мРНК FMR1 агрегация специфических белков, ведущая к митохондриальной дисфункции и клеточной гибели; повышение синтеза с того же локуса, где закодирован основной транскрипт FMR1, но ориентированной в антисмысловом направлении длинной некодирующей РНК ASFMR1/FMR4; ассоциированная с накоплением повторов, независимая от инициаторного AUG-кодона трансляция полиглицин-содержащих белковых продуктов гена FMR1 (Todd et al., 2013; Arocena et al., 2005; Loesch et al., 2011; Pastori et al., 2014).
Первый из вышеупомянутых механизмов патогенеза при наличии премутации в FMR1 – это так называемая модель “приобретения функции РНК” (“RNA gain of function”). Промоторы аллелей FMR1, ассоциированных с премутацией, не подвергаются метилированию. Было показано, что в клетках, содержащих аллели с премутацией, детектируется повышенный уровень мРНК FMR1. Такое повышение уровня транскрипции приводит к токсичности мРНК, что, в свою очередь, связано с проявлением спектра поздно манифестирующих расстройств у носителей премутации в FMR1 (Rodriguez-Revenga et al., 2009; Sherman, 2000). Предполагается, что в случае аномального повышения уровня мРНК FMR1 последняя связывается с некоторыми внутриклеточными белками, локализованными в ядре, в частности, образует комплексы с РНК-связывающими белками. С образующимися нуклеопротеиновыми агрегатами, в свою очередь, могут в дальнейшем связываться другие белки. Таким образом, избыточная мРНК FMR1 секвестрирует белки, с которыми она связывается, и препятствует их вовлечению в клеточные процессы, в которых они в норме должны быть задействованы (Greco et al., 2006; Sellier et al., 2014; Tassone et al., 2007). Это, в свою очередь, приводит к развитию синдрома FXTAS. Вышеизложенная гипотеза подтверждается тем, что при премутации гена FMR1 кодируемые им мРНК, содержащие увеличенное количество CCG-повторов, накапливаются в РНК-агрегатах в ядерных включениях, детектируемых в срезах мозга у пациентов с FXTAS (Tassone et al., 2004). Аналогичный механизм был показан при миотонической дистрофии первого типа, вызванной увеличением количества CTG повторов в 3' UTR области гена DMPK (Mankodi, Thornton, 2002).
Также было установлено, что в локусе FMR1 закодирована последовательность не только основного продукта гена FMR1, но и нескольких длинных некодирующих РНК (long noncoding RNA, lncRNA), транскрибируемых, но не транслируемых в белки. При FXS образование как минимум двух из этих lncRNA – закодированных в антисмысловом направлении транскриптов ASFMR1/FMR4 и FMR6 – подавляется эпигенетически (в силу обогащения метилированием ДНК соответствующих участков гена FMR1 и гистоновых белков хроматина). Функции FMR6 недостаточно ясны и требуют дальнейшего исследования. Однако было показано, что в норме FMR6 транскрибируется в некоторых отделах головного мозга человека; также FMR6 детектируется в периферической крови эукариот. Об ASFMR1/FMR4 известно несколько больше: эта lncRNA представляет собой ассоциированный с хроматином транскрипт. Взаимодействуя с хроматином и изменяя его состояние, ASFMR1/FMR4 регулирует экспрессию нескольких сотен генов, среди которых множество вовлечены в процессы развития нервной системы и клеточной пролиферации. Следовательно, подавление синтеза ASFMR1/FMR4 и FMR6 может вносить вклад в развитие симптомокомплекса FXS, в том числе посредством изменения экспрессии других генов в различных участках генома. Также было обнаружено, что транскрипция ASFMR1/FMR4 повышена в мозге у лиц с премутацией в FMR1, что может играть роль в развитии их фенотипа. Вместе с тем синтез FMR6 в клетках мозга у индивидуумов с премутацией в FMR1 был нарушен аналогично с картиной, которая наблюдалась для данной lncRNA у индивидуумов с полной мутацией в FMR1 (Peschansky et al., 2016; Pastori et al., 2014).
Предполагается, что токсический эффект повышения концентраций в клетках как смысловых, так и антисмысловых транскриптов, синтезируемых с локуса FMR1, при наличии аллелей FMR1, соответствующих “серой зоне” и премутации, может быть связан с развитием митохондриальной дисфункции (Loesch et al., 2011).
Однако повышенное количество мРНК, обладающее токсическим эффектом, определяется не только в случае присутствия у индивида аллеля FMR1 c премутацией, но и при наличии активной полной мутации (т.е. при отсутствии метилирования промотора FMR1, содержащего полную мутацию). Было показано, что у порядка 12–41% мужчин с FXS наблюдается мозаицизм по числу CGG-повторов и/или по наличию метилирования промотора FMR1. Мозаицизм по числу CGG-повторов наблюдается, когда в организме индивида присутствуют клетки с различными аллелями FMR1, содержащими разное количество тринуклеотидных повторов в промоторе данного гена. Наиболее частым случаем мозаицизма при FXS является присутствие в части клеток организма аллеля FMR1, соответствующего премутации, и в другой части клеток – аллеля, соответствующего полной мутации (Jiraanont et al., 2017). Такой вариант мозаицизма определяют как мозаицизм премутации/полной мутации (Rousseau et al., 1991; Nolin et al., 1994) либо мозаицизм по размеру области тринуклеотидных повторов в FMR1 (Pretto et al., 2014a). При этом аллель FMR1, несущий премутацию, может отличаться по статусу метилирования между либо внутри разных типов тканей у одного и того же индивидуума (быть как метилирован, так и транскрипционно активен), то же самое касается и аллеля FMR1, несущего полную мутацию (Field et al., 2019). Это приводит к внутритканевому и межтканевому мозаицизму. Мозаицизм по статусу метилирования при FXS также может иметь место в отсутствие у индивидуумов аллелей FMR1 с премутацией: в случае, когда некоторые клетки организма содержат метилированную форму аллеля с полной мутацией, а другие – неметилированную и транскрибируемую форму аллеля также с полной мутацией (Hagerman et al., 1994; Jiraanont et al., 2017). В нескольких случаях был описан и вариант мозаицизма, где в рамках одного организма присутствовали неметилированные аллели, соответствующие премутации и полной мутации, причем с обоих аллелей происходила продукция мРНК FMR1, а у индивидов был установлен диагноз FXTAS, основанный на клинической оценке и признаках, выявленных с помощью МРТ (Loesch et al., 2012; Pretto et al., 2013; Santa María et al., 2014).
2.3. FMRP: дефицит белка и синтез атипичных изоформ
FMRP представляет собой мультифункциональный ассоциированный с полирибосомами РНК-связывающий белок, который играет важную роль в развитии нейронов и синаптической пластичности посредством регуляции альтернативного сплайсинга мРНК, стабилизации мРНК, селективного транспорта мРНК из ядра нейронов в дендриты к синаптическим сайтам и регуляции локального синтеза определенных белков, необходимых для образования дендритов и функционирования синапсов (участвующих как в пресинаптических, так и в постсинаптических процессах передачи нервного импульса) (Antar et al., 2006; Ascano et al., 2012).
В частности, совместно с фактором ядерного экспорта NXF2 FMRP вовлечен в регуляцию стабильности мРНК NXF1 – белка, участвующего в экспорте мРНК из ядра в цитоплазму (Kim et al., 2009). Также FMRP стабилизирует мРНК DLG4/PSD-95 (postsynaptic density protein) – белка постсинаптической плотности, играющего критическую роль в синаптогенезе и синаптической пластичности (DeMarco et al., 2019). Будучи вовлеченным в механизм микроРНК-опосредованной репрессии трансляции, FMRP подавляет белковый синтез с определенных дендритных мРНК (Kenny et al., 2014). Вместе с тем FMRP играет роль активатора трансляции мРНК ряда дендритных мРНК в синапсах (Bechara et al., 2009). В недавней работе было показано, что FMRP способствует образованию миелиновой оболочки аксонов посредством локальной регуляции трансляции в олигодендроцитах (Doll et al., 2020). Также FMRP участвует в альтернативном сплайсинге собственной мРНК (Didiot et al., 2008).
Были описаны и дополнительные функции FMRP. Так, FMRP обеспечивает модуляцию ионных каналов в клетках, в частности, KCNT1 – активируемого натрием калиевого канала, в наибольшем количестве представленного в клетках мозга, но присутствующего также и во множестве других тканей (где регулирует разнообразные функции организма, включая возбудимость нейронов и высвобождение нейротрансмиттеров, частоту сердечных сокращений и сокращения гладких мышц, секрецию инсулина) (Brown et al., 2010). Также недавно было показано, что FMRP играет независимую от трансляции роль в модуляции длительности пресинаптического потенциала действия и высвобождения нейротрансмиттера через активированные кальцием калиевые каналы с большой проводимостью в возбуждающих нейронах гиппокампа и коры головного мозга (Myrick et al., 2015).
Снижение синтеза FMRP, ранее считавшееся уникальной для FXS характеристикой, также было описано у индивидуумов с премутацией (Ludwig et al., 2014).
Еще одним из ранее упомянутых (см. главу 1.2) патогенетических механизмов, который может играть роль в формировании картины нейродегенеративного заболевания у лиц с премутацией в FMR1, является RAN (repeat associated non-AUG) трансляция: независимая от AUG-кодона трансляция, ассоциированная с накоплением повторов в кодирующем белок гене. Это механизм, при котором синтез белка начинается не с канонического стартового AUG-кодона, как это происходит для большинства мРНК эукариот. Было показано, что для белков, содержащих тандемно повторяющиеся аминокислоты, в качестве альтернативного сайта инициации трансляции могут служить участки накопления тринуклеотидных повторов в регуляторных областях мРНК, кодирующих данные белки. RAN-белки могут быть синтезированы с многочисленных рамок считывания в границах одного и того же участка повторов. В результате происходит синтез и накопление целого набора белковых продуктов, нетипичных для данного гена. Предполагается, что такие белки могут нести токсический эффект (Krans et al., 2016). В частности, было продемонстрировано, что в результате реализации данного механизма у лиц с увеличенным числом CGG-повторов в промоторе FMR1 вырабатывается белок FMRpolyG (полиглицин-содержащий FMRP). Экспрессия этого белка была подтверждена в нейронах пациентов с FXTAS, полученных путем дифференцировки из индуцированных плюрипотентных стволовых клеток, и у модельных животных (Todd et al., 2013; Berman et al., 2014; Hukema et al., 2015; Sellier et al., 2017). Также процесс RAN-трансляции потенциально токсичных белков был ассоциирован с накоплением ядерных включений в нейронах у пациентов с FXTAS, что могло вносить вклад в развитие данного заболевания (Krans et al., 2016). Кроме этого, предполагается, что за развитие синдрома FXPOI отвечает действие тех же патогенетических механизмов, что и при синдроме тремора и атаксии. Было показано, что белок FMRpolyG также определяется в клетках, задействованных в фолликулогенезе, у лиц с синдромом FXPOI (Buijsen et al., 2016; Sellier et al., 2017; Friedman-Gohas et al., 2020). Таким образом, есть основание считать, что потенциально токсичная форма продукта гена FMR1, содержащего увеличенное по сравнению с нормой число CGG повторов – белок FMRpolyG – также может быть вовлечена в формирование картины нарушений репродуктивной системы у женщин с премутацией в FMR1. Подобная симптоматика не характерна для описанных женщин с полной мутацией в FMR1, при которой происходит метилирование промотора FMR1 и ингибируется транскрипция мРНК FMR1, а значит, отсутствует и синтез белка FMRpolyG. На данный момент случаев экспрессии FMR1 у женщин с полной мутацией не сообщалось, что, однако, может быть связано с трудностями дифференциации метилирования мутантного аллеля FMR1 на транскрипционно активной хромосоме X и метилирования мутантного аллеля FMR1 в результате инактивации всей хромосомы X, на которой данный аллель локализован. Диагностические методы, позволяющие решить данную проблему, были разработаны сравнительно недавно (Godler et al., 2011). Так или иначе, потенциально токсичный белок FMRpolyG может присутствовать в организме у женщин с премутацией и действовать патологически на их органы и ткани.
3. СПЕКТР РАССТРОЙСТВ FXS
Как следует из предыдущих разделов, в целом фенотипы людей с премутацией и полной мутацией в FMR1 не похожи между собой. Несмотря на это, их когнитивный, социальный и эмоционально-личностный профиль имеют много общих черт. Индивидуумы с премутацией и полной мутацией имеют схожие “пики” в когнитивном профиле, обнаруживая одинаковые сильные и слабые стороны когнитивного развития. Общей чертой также можно назвать наличие аутистических проявлений, социальную тревогу. Все это дало основание ввести термин “спектр расстройств FXS” (fragile X spectrum disorder, FXSD) (Lozano, 2014), который объединил состояния, вызванные действием двух патогенетических факторов: дефицита белка FMRP и увеличения уровня мРНК FMR1. Данный термин был разработан, чтобы подчеркнуть непрерывность клинических проявлений на всем протяжении спектра аномального количества CGG-повторов, от “серой зоны” (от 45 до 54 повторов) через состояние премутации и вплоть до полной мутации.
Анализируя весь спектр состояний, ассоциированных с мутациями гена FMR1, необходимо выделить несколько подгрупп, различающихся по патогенетическим механизмам (см. табл. 1). С точки зрения анализа отношений генотип/фенотип следует отметить, что эти подгруппы пересекаются по фенотипическим проявлениям.
Таблица 1.
Количество FMRP, уровень мРНК FMR1 в периферической крови и клинические проявления у людей с экспансией CGG-повторов Table 1. FMRP, mRNA level and phenotypic traits in people with FMR1 full mutation, premutation and mosaic forms
| FMRP (% от нормы (N)) |
мРНК (% от нормы (N)) |
Клинические проявления | |
|---|---|---|---|
| Мужчины (мальчики) с полной мутацией (число CGG-повторов в FMR1 больше 200) (Loesch et al., 2004) |
0–10% | 44–60% случаев: наблюдается неполный сайленсинг FMR1 и накопление мРНК с увеличенным количеством CCG-повторов. В остальных случаях транскрипция мРНК отсутствует (Tassone, 2001; Stoger, 2011; Baker, 2019) | IQ ≈ 40 |
| Женщины (девочки) с полной мутацией (число CGG-повторов в FMR1 больше 200) (Hall et al., 2016) |
25–50% | IQ ≈ 40–70 | |
| Мозаицизм по количеству CGG-повторовв FMR1 (Jiraanont et al., 2017a; Pretto et al., 2014) |
25–50% | >N | IQ < 70; FXTAS; FXPOI; психиатрические нарушения |
| Мозаицизм по метилированию FMR1 (Jiraanont et al., 2017a; Pretto et al., 2014) |
25–50% | N | IQ < 70 |
| Премутация (число CGG-повторов в FMR1 от 55 до 200) (Loesch, 2012) |
50–85% | >N в 3–8 раз (Tassone et al., 2000) | IQ в пределах нормы,
своеобразный когнитивный профиль, схожий с синдромом FX; FXTAS; FXPOI. Эмоционально-личностные нарушения |
| “Серая зона”
(41-54 CGG-повтора в FMR1) (Loesch et al., 2018; Hall et al., 2020; Dean et al., 2019) |
N | >N, но меньше, чем при премутации (различия стат. значимы на уровне p < 0.001) (Loesch et al., 2007) |
Повышенный риск развития паркинсонизма у мужчин и женщин, повышенный риск рецидивирующих самопроизвольных абортов |
1. Мужчины с полной мутацией в FMR1 (число тринуклеотидных повторов более 200). При таком варианте аллеля наблюдается выраженный дефицит или полное отсутствие белка FMRP. Клиническая картина достаточно однородна, расхождения наблюдаются только в степени речевого дефицита и выраженности аутистической симптоматики.
2. Женщины с полной мутацией в FMR1. Благодаря наличию второй хромосомы Х, несущей аллель дикого типа, количество белка в организме выше, чем у мужчин с полной мутацией, обычно варьирует в диапазоне 30–50% от нормы, но может принимать как более низкие, так и более высокие значения, в зависимости от особенностей инактивации хромосомы X (Hall et al., 2016; Berry-Kravis et al., 2005; Leehey et al., 2008). В соответствии с этим клиническая картина носит неоднородный характер.
3. Мозаичные формы. Схожими с предыдущим описанным вариантом по количеству белка FMRP являются мозаичные формы синдрома (составляют от 20 до 40% всех случаев синдрома FX в популяции). При FXS выделяют два вида мозаицизма – мозаицизм по числу CGG-повторов в промоторе FMR1 и мозаицизм по метилированию промотора FMR1 (Jiraanont et al., 2017a). Мозаицизм по числу CGG-повторов в данном случае проявляется в том, что в части клеток число тринуклеотидных повторов в FMR1 соответствует полной мутации, в части – премутации. Мозаицизм по метилированию предполагает, что в части клеток ген метилирован, а в части нет. Важно понимать, что в случае мозаицизма по числу CGG-повторов те клетки, в которых имеются премутационные аллели, могут накапливать мРНК FMR1, что характерно для премутации. Поэтому эти случаи, с одной стороны, не похожи на случаи полной мутации по тяжести симптомов (белок FMRP синтезируется в организме, пускай и на сниженном уровне, в силу того, что не во всех клетках полностью ингибирована его экспрессия). С другой стороны, могут возникать признаки, характерные для носителей премутации, но отсутствующие у индивидуумов с полной мутацией (в связи с токсическим эффектом повышенного уровня мРНК FMR1, транскрибируемой с содержащего премутацию гена, и с возможной трансляцией с этой мРНК токсичного белкового продукта FMRpolyG): соматические, эндокринные и нейродегенеративные заболевания. Частным случаем мозаицизма по числу CGG-повторов является описанный ранее соматический мозаицизм по мутации в FMR1. Предполагается, что количество CGG-повторов в гене FMR1 может варьировать в различных тканях (Pretto et al., 2014b).
4. Мужчины и женщины с премутацией. У людей с премутацией в FMR1 количество белка в среднем составляет 85% от нормы, колеблется в диапазоне от 50 до 100%. Избыток мРНК FMR1 может оказывать нейротоксический эффект (Loesch, 2012; Peprah et al., 2010).
5. “Серая зона”. Исследования, проведенные с участием людей с количеством повторов 44–55 (“серая зона”) немногочисленны. Есть данные, что при этом состоянии количество мРНК FMR1 статистически значимо выше, чем в норме, но ниже, чем при премутации (Loesch et al., 2007).
Таблица 1 содержит информацию о количестве белка FMRP, уровне мРНК FMR1 в клетках периферической крови и особенностях фенотипа в вышеописанных подгруппах индивидуумов. На рис. 1 графически представлены данные, касающиеся частоты появления того или иного симптома у мужчин и женщин с полной мутацией и премутацией в FMR1.
Рис. 1.
Частота (в процентах) возникновения различных клинических симптомов в группе носителей полной мутации и премутации в FMR1 (на основе данных Bailey et al., 2008).
Примечание: РАС – расстройства аутистического спектра, УО – умственная отсталость, ЗПР – задержка психического развития, FXPOI – синдром первичной овариальной недостаточности, FXTAS – синдром тремора и атаксии.
Fig. 1. Frequency (in percentage) of clinical symptoms developing in group of subjects carrier of FMR1 full mutation and premutation (based on Bailey et al., 2008).
Note: ASD – autism spectrum disorders, MR – mental retardation, DD – developmental delay, FXPOI – fragile X‑associated primary ovarian insufficiency, FXTAS – fragile X-associated tremor/ataxia syndrome.
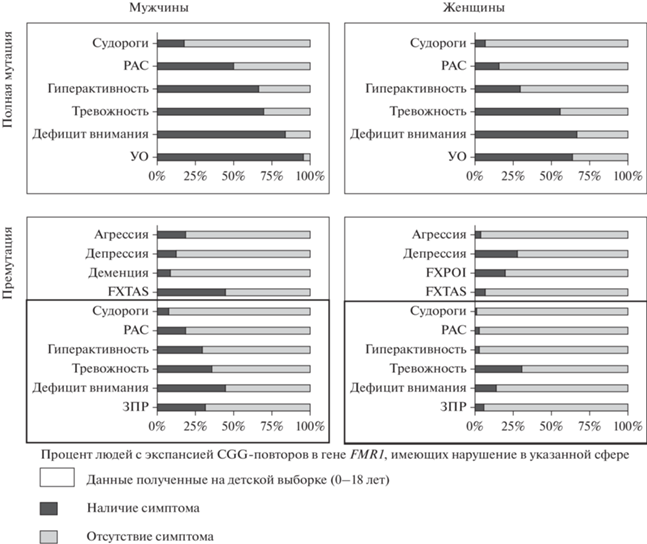
Таким образом, пересечение по фенотипу может наблюдаться: у женщин с полной мутацией, у лиц с премутацией, у мужчин с мозаицизмом по количеству повторов и по мутации в FMR1.
На рис. 1 показана частота возникновения клинических симптомов среди лиц с премутацией и полной мутацией в гене FMR1. Исходя из приведенных данных можно судить о том, что при полной мутации симптомы носят более тяжелый характер. Полиморфизм клинической картины выше среди лиц женского пола. В диапазоне премутационных изменений полиморфизм клинической картины еще выше, причем мужчины в несколько раз чаще имеют проблемы со здоровьем, чем женщины.
3.1. Связь между числом CGG-повторов в промоторе FMR1 и уровнями FMRP и мРНК FMR1
Исследования показывают, что в случае премутации FMR1 уровень экспрессии белка FMRP находится в обратной зависимости от количества CGG-повторов в промоторе данного гена (см. обзор Loesch et al., 2004). В диапазоне премутационных изменений наблюдается также положительная корреляция между количеством тринуклеотидных повторов и количеством мРНК FMR1 (Jiraanont, 2017b; Shelton, 2017). Вместе с тем существует предположение, что эта зависимость носит нелинейный характер. Так, было показано, что простая положительная корреляция между CGG-повторами и общим уровнем мРНК в крови испытуемых может быть обнаружена только при количестве повторов менее 100. Однако если построить модель, в которой используется не общий уровень мРНК, а теоретически рассчитанный уровень мРНК, транскрибируемой с мутантного аллеля (скорректировав коэффициент соотношения X‑инактивации), то оказывается, что связь между количеством повторов и уровнем мРНК усиливается и начинает превосходить ту, которая обнаруживается в диапазоне значений до 100 (Garcia-Alegria et al., 2007).
Эти результаты показывают, что существует молекулярная аномалия в экспрессии аллелей гена FMR1, несущих премутации. По мнению авторов исследования, существует некий порог в районе 100 повторов, после которого может наблюдаться выраженное увеличение (в три раза) уровня мРНК по сравнению с диапазоном с низким числом повторов (Garcia-Alegria et al., 2007). Одновременно с этим при “критическом” увеличении числа тринуклеотидных повторов в мутантном аллеле FMR1 происходит селекция клеток в пользу тех, где активна хромосома X, несущая аллель FMR1 дикого типа, что приводит к сдвигу X-инактивации в организме индивидуума. Неслучайная X-инактивация была описана и для женщин с полной мутацией в гене FMR1. Предполагается, что именно вышеописанный механизм обеспечивает вариативность экспрессии FMRP и, как следствие, широкий полиморфизм клинической картины у женщин с FXS.
3.2. Связь между числом CGG-повторов в FMR1, молекулярными механизмами патогенеза и уровнем развития когнитивных функций
В работе Д.З. Лоэш и соавторов (Loesch et al., 2004) были проанализированы результаты обследования 144 семей, члены которых имеют мутации гена FMR1 (всего 287 человек, 100 человек с полной мутацией, 95 человек с премутацией FMR1, 92 человека – норма). 87 мужчин, участвовавших в исследовании, имели полную мутацию, средние значения уровня FMRP в лимфоцитах их крови составляли 10% от количества белка в норме. 148 женщин имели премутацию в FMR1, среднее значение уровня FMRP в лимфоцитах крови у них составляло 85% от нормы. 16 человек имели мозаичную форму синдрома, уровень FMRP в лимфоцитах крови у них имел промежуточные значения (30–50%). Психологическое обследование включало тест Векслера. Помимо этого, оценивался ряд соматических черт испытуемых (включая стандартные измерения головы, лица, туловища и конечностей). Результаты исследования показали, что, во-первых, уровень FRMP в клетках крови испытуемых значимо коррелировал с уровнем их интеллекта во всех подгруппах. Причем в подгруппе мужчин эта зависимость прослеживалась в результатах всех субтестов теста Векслера, тогда как в подгруппе женщин касалась результатов выполнения только части заданий. После контроля общего балла IQ оказалось, что наиболее сильно с уровнем белка FMRP в лимфоцитах у индивидуумов коррелируют результаты тестирования их когнитивных способностей по шкалам “Запоминание цифр” (“digit span”) и “Поиск символа” (“Symbol search”) в подгруппе мужчин и “Последовательные картинки” (“Picture arrangement”) в подгруппе женщин. Эти данные свидетельствуют о том, что дефицит белка FMRP в наибольшей степени оказывает влияние на такие психические функции, как кратковременная память, внимание и скорость обработки информации. Характерно, что у лиц с аллелем гена FMR1 дикого типа также прослеживается зависимость между количеством белка FMRP в организме и когнитивными показателями, такими как IQ, рабочая память, чувствительность к стимулам магноцеллюлярного зрительного пути, восприятие движений (для обзора см. (Переверзева и др., 2015)).
Связь между количеством FMRP в лимфоцитах периферической крови и фибробластах и уровнем интеллектуального развития была также показана при сравнении данных показателей у индивидуумов с полной мутацией и с мозаицизмом по мутации/премутации и наличию/отсутствию метилирования в FMR1 (Pretto et al., 2014). Авторы исследования также указывают на достоверную корреляцию между интеллектом испытуемых и процентом клеток их крови с неметилированным аллелем FMR1.
В исследовании Д. Хессл с соавторами (Hessl et al., 2005) изучалась связь между количеством тринуклеотидных повторов в FMR1 и такими внутриклеточных показателями, как уровни FMRP и мРНК FMR1; исследование проводилось в более узкой группе – мужчин с премутацией. Результаты показали наличие связи между числом CGG-повторов в FMR1, концентрацией мРНК FMR1 в лимфоцитах и уровнем интеллекта у обследованных индивидуумов. Однако после контроля наличия синдрома тремора и атаксии (FXTAS) оказалось, что значимая корреляция наблюдается только между уровнем мРНК FMR1 в лимфоцитах (Tassone et al., 2004) и их способностью к тормозному контролю поведения (оценивался с помощью методики “Висконсинский тест сортировки карточек” – WCST). Показатели же общего интеллекта в наибольшей степени зависели от стадии развития нейродегенеративного заболевания.
В недавнем исследовании К. Ким и соавторами (Kim et al., 2019) было установлено два важных пороговых значения для уровня белка FMRP в фибробластах испытуемых: 35 и 70% от среднего значения в норме. При превышении порогового значения уровня белка, равного 70% от нормы, не наблюдалось статистически достоверной зависимости IQ индивидуумов от уровня FMRP в их фибробластах, в отличие от диапазона значений уровня белка от 35 до 50% от нормального, при которых была продемонстрирована корреляция между IQ испытуемых и уровнем FMRP. Нижняя граница нормативного диапазона IQ (85 баллов) отмечалась у пациентов с уровнем белка в их фибробластах, составлявшим 35% от средненормативного значения. При показателях уровня белка ниже этой отметки наблюдалось существенно сниженное по сравнению с нормой значение балла IQ у испытуемых.
В другом исследовании (Cohen et al., 2006) также рассматривалась связь между когнитивными нарушениями у мужчин с премутацией в FMR1 и числом тринуклеотидных повторов в данном гене. Наиболее низкие показатели интеллекта наблюдались у испытуемых, имевших высокое число CGG-повторов в промоторе FMR1. Снижение касалось показателей общего, вербального и невербального интеллекта. Причем схожий паттерн когнитивных нарушений был отмечен среди как более молодых, так и пожилых испытуемых. Таким образом, снижение когнитивных функций наблюдается у мужчин с премутацией в FMR1, число тринуклеотидных повторов в промоторе которого находится на границе значений между премутацией и полной мутацией. Иными словами, количество CGG-повторов в промоторе FMR1 можно считать предиктором когнитивных нарушений у носителей премутации в данном гене. В качестве второго фактора, влияющего на интеллект индивидуумов с премутацией в FMR1, авторы рассматривают наличие у испытуемых нейродегенеративных процессов.
В последнее десятилетие стали появляться исследования, посвященные роли в развитии синдрома FX других генетических маркеров, в частности, степени метилирования CpG-сайтов в области экзона 1 и интрона 1 гена FMR1 (в пределах FREE2 региона). У женщин с премутацией в FMR1 метилирование данной области обнаруживает значимые корреляции с тяжестью анатомических изменений в головном мозге, а также с нарушениями когнитивных функций. Так, было показано, что более высокий уровень метилирования CpG-сайта 1 гена FMR1 (локализован в экзоне 1 данного гена) в клетках периферической крови женщин с премутацией, не имеющих симптомов FXTAS, коррелировал с изменениями структуры белого вещества головного мозга, а именно – низким коэффициентом диффузии в нижних и средних ножках мозжечка (оценка производилась при помощи метода диффузионно-взвешенной визуализации). Более высокий уровень метилирования CpG-сайтов 6/7 гена FMR1 (локализованы в интроне 1) значительно коррелировал с более длительными латентными периодами просаккад (данный критерий использовался для выявления паттернов движений глаз, специфичных для нарушений исполнительных функций у женщин с премутацией в FMR1). При этом повышенный уровень метилирования CpG-сайтов 6/7 FMR1 также коррелировал с более высоким уровнем мРНК FMR1 в образцах цельной крови испытуемых. Однако, что удивительно, существенной корреляции между уровнем метилирования CpG-сайта 1 FMR1 и уровнем мРНК в образцах цельной крови испытуемых отмечено не было. Авторы предполагают, что это может быть связано с различиями в локализации границы метилирования области экзон 1/интрон 1 гена FMR1 между клетками периферической крови и клетками мозга. (Shelton et al., 2017).
Также было установлено, что степень метилирования всех 7 CpG-сайтов, расположенных в интроне 1 гена FMR1 (CpG-сайты 6/7, CpG-сайты 8/9 и CpG-сайты с 10 по 12 гена FMR1) в образцах цельной крови у женщин с премутацией значимо коррелирует с нарушениями высших исполнительных функций, а также с симптомами психических расстройств, такими как социальная тревожность (оценивалась по шкале Либовица) и нарушение регуляции эмоциональных проявлений, связанных с СДВГ (Cornish et al., 2015).
В работе Д.Е. Голдер и соавторов (Godler et al., 2012 ) было показано, что повышение степени метилирования CpG-сайтов с 10 по 12 в гене FMR1 (относительно порогового значения) в клетках крови у женщин как с премутацией, так и с полной мутацией в FMR1 со 100% чувствительностью и 98% специфичностью предсказывает низкие показатели их интеллекта, измеренные при помощи теста Векслера (IQ < 70 баллов). Аналогичным образом более высокий уровень метилирования 9 CpG-участков на границе между экзоном 1 и интроном 1 гена FMR1 (в рамках FREE2-региона), в особенности CpG-сайтов в интроне 1 FMR1, в клетках буккального эпителия, ассоциируется с более выраженным снижением уровня интеллекта (IQ, тест Векслера) у мальчиков и подростков (в возрасте от 3 до 17 лет) с синдромом FX (Arpone et al., 2018).
3.3. Связь между количеством CGG-повторов в FMR1, количеством мРНК, уровнем FMRP и симптомами РАС
Согласно различным исследованиям, до 30% индивидуумов с полной мутацией в FMR1 имеют все признаки, необходимые для постановки диагноза “аутизм”, а 50–60% удовлетворяют критериям более широкой группы психических расстройств – “расстройства аутистического спектра”, что гораздо выше, чем в среднем в популяции (Clifford et al., 2007). Это делает синдром привлекательной моделью для изучения РАС. Однако важно также понимать, что если нарушения познавательной деятельности являются обязательным признаком синдрома, напрямую следующим из дефицита белка FMRP в организме носителей полной мутации в FMR1, то РАС у данных индивидуумов наблюдаются далеко не всегда, а тяжесть проявления аутистической симптоматики широко варьирует от случая к случаю. Распространенность РАС среди людей с премутацией в FMR1 значительно выше, чем в популяции в целом, и составляет до 19% (среди мужчин), что также не позволяет считать проявление данной группы симптомов обязательным следствием действия патогенетических механизмов, запускаемых премутацией в FMR1 (Bailey et al., 2008). Интересно, что аутистическая симптоматика у лиц с FXS, в отличие от нарушений когнитивного развития, не обнаруживает значимой корреляции с количеством белка FMRP в клетках периферической крови данных индивидуумов. В некоторых исследованиях подчеркивается связь между молекулярными механизмами патогенеза, вызванными мутациями в FMR1, и особенностями социального развития лиц с FXS, однако после контроля IQ эта связь перестает быть достоверной (Loesch et al., 2007; McDuffie et al., 2010; Kover et al., 2013; Pretto et al., 2014a; Hatton et al., 2006; Hessl et al., 2001). Характерно, что также не связанными напрямую с количеством FMRP в фибробластах и клетках периферической крови при мутации в FMR1 оказываются такие показатели психической и нейрофизиологической патологии, как степень гиперактивности и частота эпилептических судорог (Pretto et al., 2014a). При мозаичных формах синдрома FX процент детей с РАС оказывается таким же, как и в группе детей с полной мутацией в FMR1, хотя уровень белка FMRP у группы с мозаичной формой значительно выше (Pretto et al., 2014a). Интересно, что, согласно данным нейровизуализационных исследований, взрослые индивидуумы с премутацией в FMR1 обнаруживают нарушения функции миндалины головного мозга (Hessl et al., 2007), которые также характерны для людей с РАС и ассоциированы с нарушениями социального развития (Baron-Cohen et al., 2000). Однако важно также учитывать, что влияние дефицита FMRP на симптоматику РАС может быть опосредованным (Budimirovic et al., 2011; Westmark et al., 2016; Ashwood et al., 2010; Sharma et al., 2010). Известно, что данный белок является регулятором трансляции множества других белков. Поэтому определенные черты развития и поведения могут быть обусловлены действием мишеней белка FMRP.
Прогресс в понимании механизмов развития РАС появился в последние годы в связи с исследованиями роли мРНК гена FMR1 при FXS. Известно, что токсический эффект накопления мРНК FMR1 ассоциирован с развитием нейродегенеративных заболеваний и овариальной недостаточности у лиц с премутацией в данном гене. Вместе с тем, как показали недавние исследования, среди лиц с полной мутацией гена FMR1 приблизительно в 44–60% случаев наблюдается неполный сайленсинг, сопровождающийся синтезом мРНК (Tassone, 2001; Stoger, 2011; Baker, 2019). Частичное либо полное ингибирование транскрипции FMR1 может зависеть от числа CGG-повторов в промоторе гена. Согласно последним данным, увеличение тринуклеотидных повторов в FMR1 до диапазона 200–400 не в 100% случаев приводит к полному метилированию гена, более вероятным порогом стабильного сайленсинга FMR1 является значение числа CGG-повторов, равное 400 (Kraan, 2019).
Оказалось, что в группе лиц мужского пола с полной мутацией в FMR1 и неполным сайленсингом (не мозаичные формы) общий балл по методике ADOS, отражающий тяжесть аутистических проявлений, был достоверно выше, чем в группе мужчин с полным сайленсингом FMR1 (Baker et al., 2019). В другом исследовании было показано, что в группе мужчин с неполным сайленсингом FMR1 уровень транскрибируемой с данного гена мРНК в клетках крови связан с уровнем аберрантного поведения испытуемых (в первую очередь, со шкалами “возбудимость” и “стереотипии”), согласно шкале ABC-C (Baker et al., 2020). Связи между уровнем мРНК FMR1 в клетках крови и интеллектом в этой группе обнаружено не было. Вместе с тем в группе детей с мозаицизмом по FMR1 уровень когнитивного развития был положительно связан с количеством мРНК данного гена. Иными словами, аутистические проявления при синдроме FX могут быть связаны с токсическим эффектом удлиненной мРНК FMR1, оверэкспрессируемой с аллеля гена FMR1, содержащего полную мутацию. Робертс с соавторами также подчеркивают, что специфика синдрома FX заключается в том, что симптомы аутизма при данной синдромальной форме тесно связаны с социальной тревожностью индивидуумов (Roberts et al., 2018). Следовательно, представляется возможным, что повышение экспрессии мРНК гена FMR1 приводит к развитию социальной тревожности у лиц с FXS, которая, в свою очередь, влечет за собой снижение их социально-коммуникативных навыков.
3.4. Связь между числом CGG-повторов в FMR1, молекулярными механизмами патогенеза и симптомами FXTAS
Количество CGG-повторов обнаруживает достоверную отрицательную корреляцию с возрастом появления двигательных нарушений, степенью моторных нарушений и возрастом смерти. Кроме того, обнаруживается достоверная положительная связь между количеством повторов и процентом нейронов и астроцитов, имеющих внутриядерные включения в сером веществе коры ГМ и гиппокампе (для обзора см. Loesch, Hagerman, 2012). Взаимосвязь между уровнем мРНК в периферической крови и клиническими проявлениями не так очевидна. Так, в исследовании Лихей (Leehey et al., 2008) не было обнаружено связи между уровнем мРНК и тяжестью симптомов FXTAS (согласно оценочной шкале FXTAS – FXTAS rating scale). Эти данные согласуются с другими исследованиями (Moore et al., 2004; Loesch et al., 2005; Jacquemont et al., 2006).
По мнению авторов, учитывая связь между уровнем мРНК FMR1 и количеством повторов, а также тот факт, что мРНК FMR1 были обнаружены во внутриядерных включениях в клетках мозга, полученные результаты могут быть объяснены расхождением между уровнем мРНК в лимфоцитах и нейронах головного мозга. Количество мРНК различно в различных отделах мозга, поэтому данный показатель, измеренный в периферической крови, не будет аккуратно отражать его экспрессию в отделах головного мозга, ассоциированных с формированием клинической симптоматики FXTAS.
Для понимания патогенетического механизма нейродегенеративных нарушений у лиц с экспансией числа тринуклеотидных повторов в FMR1 важен также анализ случаев мозаицизма по количеству повторов, при котором часть клеток в организме индивидуума содержит аллель гена с полной мутацией, а часть – с премутацией. При таком варианте также наблюдается повышение количества мРНК FMR1 и снижение экспрессии белка FMRP. Было показано, что пациенты с такой формой мозаицизма имеют повышенный риск развития неврологических двигательных симптомов (Hall et al., 2010; Loesch et al., 2012; Pretto et al., 2013, 2014). Исследования мозаицизма часто не носят репрезентативный характер, но не менее ценными оказываются описания отдельных клинических случаев, при которых наблюдается сочетание признаков FXS и FXTAS. Так, например, в исследовании Basuta (2015) описывается случай 58-летнего мужчины, генетический анализ которого выявил полную мутацию и мозаицизм по количеству повторов по FMR1 (сочетание аллелей с полной мутацией в FMR1 в мононуклеарных клетках крови и премутацией в фибробластах и сперматозоидах) при отсутствии метилирования промотора данного гена в клетках с полной мутацией. В мононуклеарных клетках крови испытуемого был повышен уровень мРНК FMR1. В фенотипе больного наиболее выделялись признаки FXTAS, нарушения памяти и исполнительного контроля. Интеллект индивидуума находился на нижней границе нормативных значений. В работе Л. Санта-Мария и соавторов (Santa Maria et al., 2014) также описан случай мозаицизма по количеству повторов, при котором наблюдалась стремительно развивающаяся форма FXTAS.
3.5. Связь между количеством CGG-повторов в FMR1, молекулярными механизмами патогенеза и синдромом первичной овариальной недостаточности
Ранее было показано, что частота возникновения синдрома первичной овариальной недостаточности (FXPOI) у женщин с премутацией в FMR1 достоверно коррелирует с количеством тринуклеотидных повторов в данном гене. Однако такая зависимость носит нелинейный характер. В исследовании Sullivan (2005) участвовало 500 женщин, имеющих нормальное и увеличенное число CGG-повторов в FMR1. Согласно результатам исследования, при числе CGG-повторов менее 40 риск развития FXPOI составляет 0.9%, при числе CGG-повторов 59–79 повышается до 5.9%, а при числе CGG-повторов 80–99 резко увеличивается до 18.6%. Однако при переходе через значение числа CGG-повторов, равное 100, этот показатель снижался до 12.5%. Эти результаты соотносятся с данными, приведенными в работе Е. Гарсия-Алегрия и соавторов (Garcia-Alegria et al., 2007), согласно которым аналогичным образом выглядит зависимость общего уровня мРНК FMR1 (определявшегося в клетках периферической крови испытуемых) от количества тринуклеотидных повторов в данном гене: наибольший уровень мРНК FMR1 детектируется у женщин с количеством CGG-повторов в диапазоне 80–100 (при отсутствии контроля X‑инактивации). В других исследованиях было показано, что средний возраст наступления менопаузы схож у женщин с премутацией в FMR1, имеющих число CGG-повторов в FMR1 в диапазоне 59–79 (48 лет) и более 100 (47.5 лет). У испытуемых с числом CGG-повторов 80–100 этот показатель был в среднем на 3 года ниже и составлял 44.9 лет (Ennis et al., 2006; Allen et al., 2007; Mailick et al., 2014). Не до конца понятным остается вопрос о том, какова роль механизма сдвига X-инактивации в описанных особенностях. С одной стороны, было показано, что при количестве CGG-повторов выше 100–120, благодаря процессу X-инактивации, уровень мРНК FMR1 (измеренный в периферической крови) перестает расти (Garcia-Alegria et al., 2007) и максимальное количество мРНК FMR1 наблюдается в диапазоне повторов 80–120. Максимальный риск развития FXPOI также наблюдается в диапазоне 80–120 повторов. Однако прямого влияния степени сдвига X‑инактивации на риски FXPOI в исследованиях обнаружено не было (Rodriguez-Revenga et al., 2009). Более того, в исследовании Майлик с соавторами (Mailick et al., 2014) было показано, что в низком и среднем диапазоне значений CGG-повторов не наблюдается корреляции между количеством повторов и степенью сдвига X-инактивации. После 100 повторов имеет место положительная связь, причем она сильнее выражена в группе женщин, имеющих FXPOI, по сравнению с теми, кто не страдает этим заболеванием. Вопрос о том, каким образом эти данные могут быть обобщены, остается открытым. Предполагается, что имеет место сложная зависимость между риском FXPOI и различными генетическими факторами (Mailick et al., 2014).
3.6. Связь между числом CGG-повторов в FMR1, молекулярными механизмами патогенеза и психиатрическими нарушениями (синдром FXAND) у лиц с FXS
Эмоционально-личностные нарушения являются характерным проявлением состояний, ассоциированных с мутациями гена FMR1. Вместе с тем исследования, посвященные отношениям генотип/фенотип в развитии симптомов FXS, затрагивающих эмоционально-личностную сферу, носят крайне противоречивый характер. Так, часть работ говорит об отсутствии связи между числом тринуклеотидных повторов в FMR1, уровнями мРНК FMR1 и белка FMRP в клетках индивидуумов с FXS и свойственными им психиатрическими диагнозами (Lachiewicz et al., 2010; Jiraanont et al., 2017b). Другие исследования указывают на линейный характер зависимости, при котором люди с более высоким числом CGG-повторов в FMR1 имеют более высокий риск развития психических заболеваний (Schneider et al., 2016; Kenna et al., 2013). Результаты третьей группы исследований доказывают обратный характер этой зависимости, при которой женщины с числом CGG-повторов в FMR1 более 100 реже демонстрируют симптомы депрессии и тревожного расстройства по сравнению с носительницами премутации в FMR1, число CGG-повторов у которых менее 100 (Loesch et al., 2015; Roberts et al., 2009a). Одним из самых репрезентативных по количеству испытуемых является исследование Loesch и соавторов (2015). В исследовании приняли участие 299 женщин в возрасте от 19 до 86 лет, имеющих количество тринуклеотидных повторов в FMR1 от 50 до 141. В качестве диагностического инструмента использовался опросник SCL-90-R, предназначенный для оценки степени выраженности паттернов различных психических расстройств и психологических признаков в рамках как психической нормы, так и патологии. Было показано, что наибольшие значения практически по всем субшкалам опросника демонстрировали женщины со средним количеством CGG-повторов (60–80), что говорит о наиболее высоком риске развития у них симптоматических комплексов различных психических заболеваний. У испытуемых с высоким числом CGG-повторов (в диапазоне 80–100), согласно результатам опросника, риск манифестации психической патологии, напротив, снижался. Эти данные представляют существенный интерес, учитывая аналогичные выводы, которые были сделаны по итогам исследования синдрома FXPOI.
В работе Cornish и соавторов (2015) изучалась связь между другим генетическим маркером – метилированием области FREE2, а именно участков CpG, расположенных в области, включающей 3'-концевой фрагмент экзона 1 и 5'-концевой фрагмент интрона 1 кодирующей последовательности гена FMR1, – и симптомами психических расстройств у женщин с премутацией в FMR1. Оказалось, что степень метилирования интрона 1 значимо коррелировала с выраженностью симптомов психических расстройств, что позволило разделить участников эксперимента на группы: высокого и низкого риска развития социальной тревожности и нарушений исполнительных функций когнитивной сферы. Оказалось, что степень сдвига X-инактивации не влияла на выраженность симптомов эмоционально-личностных нарушений. Количество CGG-повторов в FMR1 и уровень мРНК FMR1 не являлись надежными предикторами тяжести расстройств. Cornish и соавторы полагают, что степень метилирования интрона 1 FMR1 может оказаться наиболее значимым маркером развития психических нарушений, имеющим крайне высокую чувствительность (92–100%) и специфичность (90%). Характерно также, что, согласно результатам данного исследования, социальная тревожность, которая является одной из наиболее распространенных жалоб людей с премутацией, опосредована нарушениями исполнительного контроля когнитивной сферы.
Вероятно, дальнейшее изучение роли новых генетических механизмов патогенеза позволит ответить на вопросы, которые на сегодняшний день остаются неразрешенными. В первую очередь это касается связи между генетическими нарушениями у индивидуумов и обусловленными ими молекулярными механизмами развития патологии, с одной стороны, и присущими данным индивидуумам поведенческими, социальными и эмоционально-личностными нарушениями, с другой.
ВЫВОДЫ
Нарушения спектра FXS представляют собой группу заболеваний, вызванных увеличением числа тринуклеотидных повторов в промоторе гена FMR1. К основным заболеваниям спектра FMR1 относят синдромы FX, FXTAS, FXPOI, FXAND. Патогенетические механизмы состояний связаны с дефицитом экспрессии FMRP и избытком транскрипта FMR1. В статье представлено описание синдромов, описание мутаций гена FMR1 и патогенетических механизмов, ассоциированных с ними, анализ взаимосвязи между действием патогенетических механизмов и тяжестью клинических проявлений. Основываясь на представленном материале, можно сделать следующие выводы:
1. Уровень FMRP в клетках периферической крови находится в обратной зависимости от количества тринуклеотидных повторов в промоторе гена FMR1. Это утверждение касается диапазона премутационных изменений: чем выше количество повторов, тем меньше вырабатывается белка. При пересечении отметки в 200 единиц количество повторов перестает влиять на уровень синтеза FMRP, белок отсутствует либо его количество становится минимальным.
2. Уровень IQ находится в обратной зависимости от числа тринуклеотидных повторов в FMR1 и в прямой зависимости от количества FMRP. Вместе с тем не все психические функции оказываются одинаково чувствительны к дефициту белка FMRP в организме носителей мутаций в FMR1. Последствия дефицита белка FMRP в наибольшей степени затрагивают такие психические функции, как кратковременная память, внимание и скорость обработки информации.
3. Аутистическая симптоматика, в отличие от дефицита когнитивного развития, не обнаруживает значимой связи с количеством белка FMRP в клетках периферической крови испытуемых. Симптомы РАС в большей степени представлены у детей с FXS, имеющих повышенный уровень мРНК.
4. Уровень мРНК в клетках крови обнаруживает положительную корреляцию с числом тринуклеотидных повторов в промоторе гена FMR1 (в диапазоне премутационных изменений гена) у лиц мужского пола. С числом CGG-повторов в FMR1 достоверно коррелирует тяжесть и время манифестации симптомов FXTAS. Чем выше число CGG-повторов у индивидуумов, тем тяжелее у них протекает синдром тремора и атаксии, и тем раньше появляются первые симптомы. Вероятными механизмами, ответственными за развитие данного нейродегенеративного заболевания, являются механизм усиления функции РНК и RAN-трансляция FMRP.
6. Частота возникновения FXPOI достоверно коррелирует с количеством CGG-повторов в FMR1. Наиболее вероятной причиной развития синдрома FXPOI является накопление мРНК, кодируемой геном FMR1. Количество повторов в диапазоне 90–110 ассоциировано с более ранним возрастом наступления менопаузы. Предполагается, что за развитие синдрома FXPOI могут отвечать те же механизмы, что и за развитие синдрома FXTAS – усиление функции РНК и RAN-трансляция.
7. С выраженностью симптомов психических заболеваний надежно коррелирует степень метилирования CpG-участков интрона 1 кодирующей области гена FMR1. Этот показатель оказался весьма надежным критерием (100% чувствительность и 98% специфичность), позволяющим разделить испытуемых на группы высокого и низкого риска развития психо-эмоциональных проблем и нарушений исполнительных функций. Метилирование данного участка также надежно коррелирует с анатомическими изменениями головного мозга, ассоциированными с нарушениями исполнительных функций когнитивной сферы.
8. Степень метилирования интрона 1 гена FMR1 также ассоциирована с уровнем интеллекта у женщин и мужчин с полной мутацией в FMR1.
Список литературы
Переверзева Д.С., Данилина К.К., Горбачевская Н.Л. Общие и специфические механизмы нарушения развития зрительной когнитивной функции у лиц с дефицитом белка FMRP. Журн. высш. нервн. деят. им. И.П. Павлова. 2015. 65(3): 259–270.
Переверзева Д.С., Тюшкевич С.А., Горбачевская Н.Л., Мамохина У.А., Данилина К.К. Гетерогенность клинической картины при синдромах, ассоциированных с динамическими мутациями гена FMR1. Журнал неврологии и психиатрии им. С.С. Корсакова. 2019. 119(7): 103–111.
Тюшкевич С.А., Воинова В.Ю., Юров И.Ю., Горбачевская Н.Л. Характер когнитивных нарушений у детей с синдромом умственной отсталости, сцепленной с ломкой хромосомой Х. Дефектология. 2010. 3: 29–37.
Юдкин Д.В., Лемская Н.А., Грищенко И.В., Дольский А.А. Изменение состава хроматина при экспансии тринуклеотидного повтора CGG в гене FMR1. Молекулярная биология. 2015. 49(2): 205–211.
Abitbol M., Menini C., Delezoide A.-L., Rhyner T., Vekemans M., Mallet J. Nucleus basalis magnocellularis and hippocampus are the major sites of FMR-1 expression in the human fetal brain. Nature. Genet. 1993. 4: 147–153.
Allen E.G., Sullivan A.K., Marcus M., Small C., Dominguez C., Epstein M.P., Charen K., He W., Taylor K.C., Sherman S.L. Examination of reproductive aging milestones among women who carry the FMR1 premutation. Hum. Reprod. 2007. 22(8): 2142–2152.
Antar L.N., Li C., Zhang H., Carroll R.C., Bassell G.J. Local functions for FMRP in axon growth cone motility and activity-dependent regulation of filopodia and spine synapses. Mol. Cell. Neurosci. 2006. 32: 37–48.
Arocena D.G., Iwahashi C.K., Won N., Beilina A., Ludwig A.L., Tassone F., Schwartz P.H., Hagerman P.J. Induction of inclusion formation and disruption of lamin A/C structure by premutation CGG-repeat RNA in human cultured neural cells. Hum. Mol. Genet. 2005. 14: 3661–3671.
Arpone M., Baker E.K., Bretherton L., Bui M., Li X., Whitaker S., Dissanayake C., Cohen J., Hickerton C., Rogers C., Field M., Elliott J., Aliaga S.M., Ling L., Francis D., Hearps S., Hunter M.F., Amor D.J., Godler D.E. Intragenic DNA methylation in buccal epithelial cells and intellectual functioning in a paediatric cohort of males with fragile X. Sci. Rep. 2018. 8(1): 3644.
Ascano M.Jr., Mukherjee N., Bandaru P., Miller J.B., Nusbaum J.D., Corcoran D.L., Langlois C., Munschauer M., Dewell S., Hafner M., Williams Z., Ohler U., Tuschl T. FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature. 2012. 492: 382–386.
Ashwood P., Nguyen D.V., Hessl D., Hagerman R.J., Tassone F. Plasma cytokine profiles in Fragile X subjects: is there a role for cytokines in the pathogenesis? Brain. Behav. Immun. 2010. 24(6): 898–902.
Bailey D.B.Jr., Raspa M., Olmsted M., Holiday D.B. Co-occurring conditions associated with FMR1 gene variations: findings from a national parent survey. Am. J. Med. Genet. 2008. 146A(16): 2060–2069.
Baker E.K., Arpone M., Aliaga S.M., Bretherton L., Kraan C.M., Bui M., Slater H.R., Ling L., Francis D., Hunter M.F., Elliott J., Rogers C., Field M., Cohen J., Cornish K., Santa Maria L., Faundes V., Curotto B., Morales P., Trigo C., Salas I., Alliende A.M., Amor D.J., Godler D.E. Incomplete silencing of full mutation alleles in males with fragile X syndrome is associated with autistic features. Mol. Autism. 2019. 3: 10–21.
Baker E.K., Arpone M., Kraan C., Bui M., Rogers C., Field M., Bretherton L., Ling L., Ure A., Cohen J., Hunter M.F., Santa María L., Faundes V., Curotto B., Morales P., Trigo C., Salas I., Alliende A., Amor D.J., Godler D.E. FMR1 mRNA from full mutation alleles is associated with ABC-CFX scores in males with fragile X syndrome. Sci. Rep. 2020. 10(1): 11701.
Baron-Cohen S., Ring H.A., Bullmore E.T., Wheelwright S., Ashwin C., Williams S.C. The amygdala theory of autism. Neurosci. Biobehav. Rev. 2000. 24(3): 355–364.
Basuta K., Schneider A., Gane L., Polussa J., Woodruff B., Pretto D., Hagerman R., Tassone F. High functioning male with fragile X syndrome and fragile X-associated tremor/ataxia syndrome. Am. J. Med. Genet. 2015. 167A (9): 2154–2161.
Bechara E.G., Didiot M.C., Melko M., Davidovic L., Bensaid M., Martin P., Castets M., Pognonec P., Khandjian E.W., Moine H., Bardoni B. A novel function for fragile X mental retardation protein in translational activation. PLoS. Biol. 2009. 7: E16–E16.
Berman R.F., Buijsen R.A., Usdin K., Pintado E., Kooy F., Pretto D., Pessah I.N., Nelson D.L., Zalewski Z., Charlet-Bergeurand N., Willemsen R., Hukema R.K. Mouse models of the fragile X premutation and fragile X-associated tremor/ataxia syndrome. J. Neurodev. Disord. 2014. 6(1): 25.
Berry-Kravis E., Potanos K., Weinberg D., Zhou L., Goetz C.G. Fragile X-associated tremor/ataxia syndrome in sisters related to X-inactivation. Ann. Neurol. 2005. 57: 144–147.
Brown M.R., Kronengold J., Gazula V.R., Chen Y., Strumbos J.G., Sigworth F.J., Navaratnam D., Kaczmarek L.K. Fragile X mental retardation protein controls gating of the sodium-activated potassium channel Slack. Nat. Neurosci. 2010. 13: 819–821.
Budimirovic D.B., Kaufmann W.E. What can we learn about autism from studying fragile X syndrome? Dev. Neurosci. 2011. 33(5): 379–394.
Budimirovic D.B, Subramanian M. Neurobiology of Autism and Intellectual Disability: fragile X Syndrome. 2 ed. In: Johnston M., Adams H., Fatemi A., editors. London: Oxford University Press; 2016.
Buijsen R.A., Visser J.A., Kramer P., Severijnen E.A., Gearing M., Charlet-Berguerand N., Sherman S.L., Berman R.F., Willemsen R., Hukema R.K. Presence of inclusions positive for polyglycine containing protein, FMRpolyG, indicates that repeat-associated non-AUG translation plays a role in fragile X-associated primary ovarian insufficiency. Hum. Reprod. 2016. 31(1): 158–168.
Clifford S., Dissanayake C., Bui Q.M., Huggins R., Taylor A.K., Loesch D.Z. Autism spectrum phenotype in males and females with fragile X full mutation and premutation. Journal of Autism and Developmental Disorders. 2007. 37(4): 738–747.
Cohen S., Masyn K., Adams J., Hessl D., Rivera S., Tassone F., Brunberg J., DeCarli C., Zhang L., Cogswell J., Loesch D., Leehey M., Grigsby J., Hagerman P.J., Hagerman R. Molecular and imaging correlates of the fragile X-associated tremor/ataxia syndrome. Neurology. 2006. 67(8): 1426–1431.
Colak D., Zaninovic N., Cohen M.S., Rosenwaks Z., Yang W.Y., Gerhardt J., Disney M.D., Jaffrey S.R. Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome. Science. 2014. 343(6174): 1002–1005.
Cornish K.M., Kogan C.S., Li L., Turk J., Jacquemont S., Hagerman R.J. Lifespan changes in working memory in fragile X premutation males. Brain. Cogn. 2009. 69(3): 551–558.
Cornish K.M., Kraan C.M., Bui Q.M., Bellgrove M.A., Metcalfe S.A., Trollor J.N., Hocking D.R., Slater H.R., Inaba Y., Li X., Archibald A.D., Turbitt E., Cohen J., Godler D.E. Novel methylation markers of the dysexecutive-psychiatric phenotype in FMR1 premutation women. Neurology. 2015. 84(16): 1631–1638.
de Vries B.B., Wiegers A.M., Smits A.P., Mohkamsing S., Duivenvoorden H.J., Fryns J.P., Curfs L.M., Halley D.J., Oostra B.A., van den Ouweland A.M., Nier- meijer M.F. Mental status of females with an FMR1 gene full mutation. Am. J. Hum. Genet. 1996. 58(5): 1025–1032.
Dean D.D., Agarwal S., Muthuswamy S. Defining the role of FMR1 gene in unexplained recurrent spontaneous abortion. J Assist Reprod Genet. 2019. 36(11): 2245–2250.
DeMarco B., Stefanovic S., Williams A., Moss K.R., Anderson B.R., Bassell G.J., Mihailescu M.R. FMRP – G-quadruplex mRNA - miR-125a interactions: Implications for miR-125a mediated translation regulation of PSD-95 mRNA. PLoS One. 2019. 14(5): e0217275.
Didiot M.C., Tian Z., Schaeffer C., Subramanian M., Mandel J.L., Moine H. The G-quartet containing FMRP binding site in FMR1 mRNA is a potent exonic splicing enhancer. Nucleic. Acids. Res. 2008. 36: 4902–4912.
Doll C.A., Yergert K.M., Appel B.H. The RNA binding protein fragile X mental retardation protein promotes myelin sheath growth Glia. 2020. 68(3): 495–508.
Drouin R., Angers M., Dallaire N., Rose T.M., Khandjian W., Rousseau F. Structural and functional characterization of the human FMR1 promoter reveals similarities with the hnRNP-A2 promoter region. Hum. Mol. Genet. 1997. 6: 2051–2060.
Ennis S., Ward D., Murray A. Nonlinear association between CGG repeat number and age of menopause in FMR1 premutation carriers. Eur. J. Hum. Genet. 2006. 14(2): 253–255.
Farzin F., Perry H., Hessl D., Loesch D., Cohen J., Bacalman S., Gane L., Tassone F., Hagerman P., Hagerman R. Autism spectrum disorders and attention-deficit/hyperactivity disorder in boys with the fragile X premutation. J Dev Behav Pediatr. 2006. 27(2 Suppl): 137–144. PMID: 16685180.https://doi.org/10.1097/00004703-200604002-00012
Field M., Dudding-Byth T., Arpone M., Baker E.K., Aliaga S.M., Rogers C., Hickerton C., Francis D., Phelan D.G., Palmer E.E., Amor D.J., Slater H., Bretherton L., Ling L., Godler D.E. Significantly Elevated FMR1 mRNA and Mosaicism for Methylated Premutation and Full Mutation Alleles in Two Brothers with Autism Features Referred for Fragile X Testing. Int J Mol Sci. 2019. 20(16): 3907.
Friedman-Gohas M., Elizur S.E., Dratviman-Storobinsky O., Aizer A., Haas J., Raanani H., Orvieto R., Cohen Y. FMRpolyG accumulates in FMR1 premutation granulosa cells. J Ovarian Res. 2020. 13(1): 22.
Gallego P.K., Burris J.L., Rivera S.M. Visual motion processing deficits in infants with the fragile X premutation. J. Neurodev Disord. 2014. 6(1): 29. https://doi.org/10.1186/1866-1955-6-29
Garcia-Alegria E., Ibáñez B., Mínguez M., Poch M., Valiente A., Sanz-Parra A., Martinez-Bouzas C., Beristain E., Tejada M.I. Analysis of FMR1 gene expression in female premutation carriers using robust segmented linear regression models. RNA. 2007. 13(5): 756–762.
Godler D.E., Slater H.R., Bui Q.M., Ono M., Gehling F., Francis D., Amor D.J., Hopper J.L., Hagerman R., Loesch D.Z. FMR1 intron 1 methylation predicts FMRP expression in blood of female carriers of expanded FMR1 alleles. J. Mol. Diagn. 2011. 13(5): 528–536.
Godler D.E., Slater H.R., Bui Q.M., Storey E., Ono M.Y., Gehling F., Inaba Y., Francis D., Hopper J.L., Kinsella G., Amor D.J., Hagerman R.J., Loesch D.Z. Fragile X mental retardation 1 (FMR1) intron 1 methylation in blood predicts verbal cognitive impairment in female carriers of expanded FMR1 alleles: evidence from a pilot study. Clin. Chem. 2012. 58(3): 590–598.
Godler D.E., Tassone F., Loesch D.Z., Taylor A.K., Gehling F., Hagerman R.J., Burgess T., Ganesamoorthy D., Hennerich D., Gordon L., Evans A., Choo K.H., Slater H.R. Methylation of novel markers of fragile X alleles is inversely correlated with FMRP expression and FMR1 activation ratio. Hum. Mol. Genet. 2010. 19(8): 1618–1632.
Godler D.E. Treatment and diagnosis of epigenetic disorders and conditions. Google Patents; 2012.
Greco C.M., Berman R.F., Martin R.M., Tassone F., Schwartz P.H., Chang A., Trapp B.D., Iwahashi C., Brunberg J., Grigsby J., Hessl D., Becker E.J., Papazian J., Leehey M.A., Hagerman R.J., Hagerman P.J. Neuropathology of fragile X-associated tremor/ataxia syndrome (FXTAS). Brain. 2006. 129 (Pt 1): 243–255.
Hagerman P.J., Hagerman R.J. The fragile-X premutation: a maturing perspective. Am. J. Hum. Genet. 2004. 74(5): 805–816.
Hagerman R.J., Berry-Kravis E., Hazlett H.C., Bailey D.B. Jr., Moine H., Kooy R.F., Tassone F., Gantois I., Sonenberg N., Mandel J.L., Hagerman P.J. Fragile X syndrome. Nat. Rev. Dis. Primers. 2017. 3: 17065.
Hagerman R.J., Hull C.E., Safanda J.F., Carpenter I., Staley L.W., O’Connor R.A., Seydel C., Mazzocco M.M., Snow K., Thibodeau S.N., Kuhl D., Nelson D.L., Caskey C.Th., Taylor A.K. High functioning fragile X males: demonstration of an unmethylated fully expanded FMR-1 mutation associated with protein expression. Am J Med Genet. 1994. 51(4): 298–308.
Hagerman R.J., Protic D., Rajaratnam A., Salcedo-Arellano M.J., Aydin E.Y., Schneider A. Fragile X-Associated Neuropsychiatric Disorders (FXAND). Frontiers in psychiatry. 2018. 9: 564.
Hall D., Pickler L., Riley K., Tassone F., Hagerman R. Parkinsonism and cognitive decline in a fragile X mosaic male. Mov. Disord. 2010. 25(10): 1523–1524.
Hall D.A., Robertson-Dick E.E., O’Keefe J.A., Hadd A.G., Zhou L., Berry-Kravis E. X-inactivation in the clinical phenotype of fragile X premutation carrier sisters. Neurology. Genetics. 2016. 2(1): e45.
Hall D.A., Nag S., Ouyang B., Bennett D.A., Liu Y., Ali A., Zhou L., Berry-Kravis E. Fragile X Gray Zone Alleles Are Associated With Signs of Parkinsonism and Earlier Death. Mov. Disord. 2020. 35(8): 1448–1456.
Hashimoto R.I., Backer K.C., Tassone F., Randi J., Hagerman R.J., Rivera S.M. An fMRI study of the prefrontal activity during the performance of a work- ing memory task in premutation carriers of the fragile X mental retardation 1 gene with and without fragile X-associated tremor/ataxia syndrome (FX-TAS). Journal of Psychiatric Research. 2010. 45(1): 36–43.
Hatton D.D., Sideris J., Skinner M., Mankowski J., Bailey D.B. Jr, Roberts J., Mirrett P. Autistic behavior in children with fragile X syndrome: prevalence, stability, and the impact of FMRP. Am. J. Med. Genet. A. 2006. 140A(17): 1804–1813.
Heine-Suñer D., Torres-Juan L., Morlà M., Busquets X., Barceló F., Picó G., Bonilla L., Govea N., Bernués M., Rosell J. Fragile-X syndrome and skewed X-chromosome inactivation within a family: a female member with complete inactivation of the functional X chromosome. Am J Med Genet A. 2003. 122A(2): 108–114.
Hessl D., Dyer-Friedman J., Glaser B., Wisbeck J., Barajas R.G., Taylor A., Reiss A.L. The influence of environmental and genetic factors on behavior problems and autistic symptoms in boys and girls with fragile X syndrome. Pediatrics. 2001. 108(5): E88.
Hessl D., Tassone F., Loesch D.Z., Berry-Kravis E., Leehey M.A., Gane L.W., Barbato I., Rice C., Gould E., Hall D.A., Grigsby J., Wegelin J.A., Harris S., Lewin F., Weinberg D., Hagerman P.J., Hagerman R.J. Abnormal elevation of FMR1 mRNA is associated with psychological symptoms in individuals with the fragile X premutation. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2005. 139B(1): 115–121.
Hessl D., Nguyen D.V., Green C., Chavez A., Tassone F., Hagerman R. J., Senturk D., Schneider A., Lightbody A., Reiss A.L., Hall S. A solution to limitations of cognitive testing in children with intellectual disabilities: the case of fragile X syndrome. Journal of neurodevelopmental disorders. 2009. 1(1): 33–45.
Hinds H.L., Ashley C.T., Sutcliffe J.S., Nelson D.L., Warren S.T., Housman D.E., Schalling M. Tissue specific expression of FMR-1 provides evidence for a functional role in fragile X syndrome. Nat. Genet. 1993. 3(1): 36–43.
Hukema R.K., Buijsen R.A., Schonewille M., Raske C., Severijnen L.A., Nieuwenhuizen-Bakker I., Verhagen R.F., van Dessel L., Maas A., Charlet-Berguerand N., De Zeeuw C.I., Hagerman P.J., Berman R.F., Willemsen R. Reversibility of neuropathology and motor deficits in an inducible mouse model for FXTAS. Hum. Mol. Genet. 2015. 24(17): 4948–4957.
Jacquemont S., Hagerman R.J., Leehey M.A., Hall D.A., Levine R.A., Brunberg J.A., Zhang L., Jardini T., Gane L.W., Harris S.W., Herman K., Grigsby J., Greco C.M., Berry-Kravis E., Tassone F., Hagerman P.J. Penetrance of the fragile X-associated tremor/ataxia syndrome in a premutation carrier population. JAMA. 2004. 291(4): 460–469.
Jacquemont S., Leehey M.A., Hagerman R.J., Beckett L.A., Hagerman P.J. Size bias of fragile X premutation alleles in late-onset movement disorders. J. Med. Genet. 2006. 43(10): 804–809.
Jalnapurkar I., Rafika N., Tassone F., Hagerman R. Immune mediated disorders in women with a fragile X expansion and FXTAS. Am. J. Med. Genet. A. 2015. 167A(1): 190–7.
Jiraanont P., Kumar M., Tang H.T., Espinal G., Hagerman P.J., Hagerman R.J., Chutabhakdikul N., Tassone F. Size and methylation mosaicism in males with Fragile X syndrome. Expert. Rev. Mol. Diagn. 2017a. 17(11): 1023–1032.
Jiraanont P., Sweha S.R., AlOlaby R.R., Silva M., Tang H.T., Durbin-Johnson B., Schneider A., Espinal G.M., Hagerman P.J., Rivera S.M., Hessl D., Hagerman R.J., Chutabhakdikul N., Tassone F. Clinical and molecular correlates in fragile X premutation females. eNeurologicalSci. 2017b. 7: 49–56.
Kenna H.A., Tartter M., Hall S.S. High rates of comorbid depressive and anxiety disorders among women with premutation of the FMR1 gene. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2013. 162(8): 872–878.
Kenny P.J., Zhou H., Kim M., Skariah G., Khetani R.S., Drnevich J., Arcila M.L., Kosik K.S., Ceman S. MOV10 and FMRP regulate AGO2 association with microRNA recognition elements. Cell Rep. 2014. 9: 1729–1741.
Kim K., Hessl D., Randol J.L., Espinal G.M., Schneider A., Protic D., Aydin E.Y., Hagerman R.J., Hagerman P.J. Association between IQ and FMR1 protein (FMRP) across the spectrum of CGG repeat expansions. PLoS One. 2019. 14(12): e0226811.
Kim M., Bellini M., Ceman S. Fragile X mental retardation protein FMRP binds mRNAs in the nucleus. Mol. Cell. Biol. 2009. 29: 214–228.
Kirchgessner C.U., Warren S.T., Willard H.F. X inactivation of the FMR1 fragile X mental retardation gene. Journal of medical genetics. 1995. 32(12): 925–929.
Kover S.T., Pierpont E.I., Kim J.S., Brown W.T., Abbeduto L. A neurodevelopmental perspective on the acquisition of nonverbal cognitive skills in adolescents with fragile X syndrome. Dev. Neuropsychol. 2013. 38(7): 445–460.
Kraan C.M., Godler D.E., Amor D.J. Epigenetics of fragile X syndrome and fragile X-related disorders. Dev. Med. Child. Neurol. 2019. 61(2): 121–127.
Krans A., Kearse M.G., Todd P.K. Repeat-associated nonAUG translation from antisense CCG repeats in fragile X tremor/ataxia syndrome. Ann Neurol. 2016. 80: 871–881.
Kumari D., Usdin K. Interaction of the transcription factors USF1, USF2, and α-Pal/Nrf-1 with the FMR1 promoter. Implications for fragile X mental retardation syndrome. J. Biol. Chem. 2001. 276: 4357–4364.
Lachiewicz A., Dawson D., Spiridigliozzi G. Indicators of anxiety and depression in women with the fragile X premutation: assessment of a clinical sample. J. Intellect. Disabil. Res. 2010. 54(7): 597–610.
Leehey M.A., Berry-Kravis E., Goetz C.G., Zhang L., Hall D.A., Li L., Rice C.D., Lara R., Cogswell J., Reynolds A., Gane L., Jacquemont S., Tassone F., Grigsby J., Hagerman R.J., Hagerman P.J. FMR1 CGG repeat length predicts motor dysfunction in premutation carriers. Neurology. 2008. 70(16 Pt 2): 1397–402.
Loesch D.Z., Bui M.Q., Hammersley E. Psychological status in female carriers of premutation FMR1 allele showing a complex relationship with the size of CGG expansion. Clin. Genet. 2015. 87(2): 173–178.
Loesch D.Z., Bui Q.M., Dissanayake C., Clifford S., Gould E., Bulhak-Paterson D., Tassone F., Tay-lor A.K., Hessl D., Hagerman R., Huggins R.M. Molecular and cognitive predictors of the continuum of autistic behaviours in fragile X. Neurosci. Biobehav. Rev. 2007. 31(3): 315–326.
Loesch D.Z., Godler D.E., Evans A., Bui Q.M., Gehling F., Kotschet K.E., Trost N., Storey E., Stimpson P., Kinsella G., Francis D., Thorburn D.R., Venn A., Slater H.R., Horne M. Evidence for the toxicity of bidirectional transcripts and mitochondrial dysfunction in blood associated with small CGG expansions in the FMR1 gene in patients with parkinsonism. Genet. Med. 2011. 13: 392–399.
Loesch D.Z., Huggins R.M., Hagerman R.J. Phenotypic variation and FMRP levels in fragile X. Ment. Retard. Dev. Disabil. Res. Rev. 2004. 10(1): 31–41.
Loesch D.Z., Sherwell S., Kinsella G., Tassone F., Taylor A., Amor D., Sung S., Evans A. Fragile X-associated tremor/ataxia phenotype in a male carrier of unmethylated full mutation in the FMR1 gene. Clin. Genet. 2012. 82(1): 88–92.
Loesch D.Z., Tassone F., Mellick G.D., Horne M., Rubio J.P., Bui M.Q., Francis D., Storey E. Evidence for the role of FMR1 gray zone alleles as a risk factor for parkinsonism in females. Mov Disord. 2018. 33(7): 1178–1181.
Loesch D.Z., Churchyard A., Brotchie P., Marot M., Tassone F. Evidence for, and a spectrum of, neurological involvement in carriers of the fragile X premutation: FXTAS and beyond. Clin Genet. 2005. 67: 412–417.
Loesch D., Hagerman R. Unstable mutations in the FMR1 gene and the phenotypes. Adv Exp Med Biol. 2012. 769: 78–114. https://doi.org/10.1007/978-1-4614-5434-2_6
Lozano R., Rosero C., Hagerman R. Fragile X spectrum disorders. Intractable & Rare Diseases Research. 2014. 3(4): 134–146.
Ludwig A.L., Espinal G.M., Pretto D., Jamal A.L., Arque G., Tassone F., Berman R.F., Hagerman P.J. CNS expression of murine fragile X protein (FMRP) as a function of CGG-repeat size. Hum. Mol. Genet. 2014. 23: 3228–3238.
Mailick M.R., Hong J., Greenberg J., Smith L., Sherman S. Curvilinear association of CGG repeats and age at menopause in women with FMR1 premutation expansions. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2014. 165B(8): 705–711.
Mankodi A., Thornton C.A. Myotonic syndromes. Curr. Opin.Neurol. 2002. 15: 545–552.
McDuffie A., Abbeduto L., Lewis P., Kover S., Kim J.S., Weber A., Brown W.T. Autism spectrum disorder in children and adolescents with fragile X syndrome: within-syndrome differences and age-related changes. Am. J. Intellect. Dev. Disabil. 2010. 115(4): 307–326.
Moore C.J., Daly E.M., Schmitz N., Tassone F., Tysoe C., Hagerman R.J., Hagerman P.J., Morris R.G., Murphy K.C., Murphy D.G. A neuropsychological investigation of male premutation carriers of fragile X syndrome. Neuropsychologia. 2004. 42: 1934–1947.
Myrick L.K., Deng P.Y., Hashimoto H., Oh Y.M., Cho Y., Poidevin M.J., Suhl J.A., Visootsak J., Cavalli V., Jin P., Cheng X., Warren S.T., Klyachko V.A. Independent role for presynaptic FMRP revealed by an FMR1 missense mutation associated with intellectual disability and seizures. Proc. Natl. Acad. Sci. U.S.A. 2015. 112: 949–956.
Naumann A., Hochstein N., Weber S., Fanning E., Doerfler W. A distinct DNA-methylation boundary in the 5′-upstream sequence of the FMR1 promoter binds nuclear proteins and is lost in fragile X syndrome. Am. J. Hum. Genet. 2009. 85: 606–616.
Nelson L.M., Covington S.N., Rebar R.W. An update: spontaneous premature ovarian failure is not an early menopause. Fertil. Steril. 2005. 283: 1327–1332.
Nolin S.L., Glicksman A., Houck G.E. Jr, Brown W.T., Dobkin C.S. Mosaicism in fragile X affected males. Am. J. Med. Genet. 1994. 51(4): 509–512.
Pastori C., Peschansky V.J., Barbouth D., Mehta A., Silva J.P., Wahlestedt C. Comprehensive analysis of the transcriptional landscape of the human FMR1 gene reveals two new long noncoding RNAs differentially expressed in Fragile X syndrome and Fragile X-associated tremor/ataxia syndrome. Hum. Genet 2014. 133: 59–67.
Peprah E., He W., Allen E., Oliver T., Boyne A., Sherman S.L. Examination of FMR1 transcript and protein levels among 74 premutation carriers. J. Hum. Genet. 2010. 55(1): 66–68.
Peschansky V.J., Pastori C., Zeier Z., Wentzel K., Velmeshev D., Magistri M., Silva J.P., Wahlestedt C. The long noncoding RNA FMR4 promotes proliferation of human neural precursor cells and epigenetic regulation of gene expression in trans. Mol. Cell. Neurosci. 2016. 74: 49–57.
Pietrobono R., Pomponi M.G., Tabolacci E., Oostra B., Chiurazzi P., Neri G. Quantitative analysis of DNA demethylation and transcriptional reactivation of the FMR1 gene in fragile X cells treated with 5‑azadeoxycytidine. Nucleic. Acids. Res. 2002. 30: 3278–3285.
Pretto D., Yrigollen C.M., Tang H.T., Williamson J., Espinal G., Iwahashi C.K., Durbin-Johnson B., Hagerman R.J., Hagerman P.J., Tassone F. Clinical and molecular implications of mosaicism in FMR1 full mutations. Front Genet. 2014a. 5: 318.
Pretto D.I., Hunsaker M.R., Cunningham C.L., Greco C.M., Hagerman R.J., Noctor S.C., Hall D.A., Hagerman P.J., Tassone F. Intranuclear inclusions in a fragile X mosaic male. Transl. Neurodegener. 2013. 2(1): 10.
Pretto D.I., Mendoza-Morales G., Lo J., Cao R., Hadd A., Latham G.J., Durbin-Johnson B., Hagerman R., Tassone F. CGG allele size somatic mosaicism and methylation in FMR1 premutation alleles. J. Med. Genet. 2014b. 51(5): 309–318.
Rajaratnam A., Shergill J., Salcedo-Arellano M., Saldarriaga W., Duan X., Hagerman R. Fragile X syndrome and fragile X-associated disorders. F1000Res. 2017. 6: 2112.
Raspa M., Wylie A., Wheeler A.C., Kolacz J., Edwards A., Heilman K., Porges S.W. Sensory Difficulties in Children With an FMR1 Premutation. Front Genet. 2018 Aug 28; 9: 351. PMID: 30233641; PMCID: PMC6127619.https://doi.org/10.3389/fgene.2018.00351
Roberts J.E., Bailey D.B., Mankowski J. Mood and anxiety disorders in females with the FMR1 premutation. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2009a. 150(1): 130–139.
Roberts J.E., Ezell J.E., Fairchild A.J., Klusek J., Thurman A.J., McDuffie A., Abbeduto L. Biobehavioral composite of social aspects of anxiety in young adults with fragile X syndrome contrasted to autism spectrum disorder. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2018. 177(7): 665–675.
Rodriguez-Revenga L., Madrigal I., Badenas C., Xunclà M., Jiménez L., Milà M. Premature ovarian failure and fragile X female premutation carriers: no evidence for a skewed X-chromosome inactivation pattern. Menopause. 2009. 16(5): 944–949.
Rodriguez-Revenga L., Madrigal I., Pagonabarraga J., Xunclà M., Badenas C., Kulisevsky J., Gomez B., Milà M. Penetrance of FMR1 premutation associated pathologies in fragile X syndrome families. Eur. J. Hum. Genet. 2009. 17(10): 1359–1362.
Rousseau F., Heitz D., Biancalana V., Blumenfeld S., Kretz C., Boué J., Tommerup N., Van Der Hagen C., DeLozier-Blanchet C., Croquette M.F. Direct diagnosis by DNA analysis of the fragile X syndrome of mental retardation. N. Engl. J. Med. 1991. 325(24): 1673–1681.
Salcedo-Arellano M.J., Dufour B., McLennan Y., Martinez-Cerdeno V., Hagerman R. Fragile X syndrome and associated disorders: Clinical aspects and pathology. Neurobiol Dis. 2020 Mar; 136: 104740.
Santa María L., Pugin A., Alliende M.A., Aliaga S., Curotto B., Aravena T., Tang H.T., Mendoza-Morales G., Hagerman R., Tassone F. FXTAS in an unmethylated mosaic male with fragile X syndrome from Chile. Clin. Genet. 2014. 86(4): 378–382.
Schneider A., Johnston C., Tassone F. Broad autism spectrum and obsessive–compulsive symptoms in adults with the fragile X premutation. Clin. Neuropsychol. 2016. 30(6): 929–943.
Sellier C., Buijsen RAM, He F., Natla S., Jung L., Tropel P., Gaucherot A., Jacobs H., Meziane H., Vincent A., Champy M.F., Sorg T., Pavlovic G., Wattenhofer-Donze M., Birling M.C., Oulad-Abdelghani M., Eberling P., Ruffenach F., Joint M., Anheim M., Martinez-Cerdeno V., Tassone F., Willemsen R., Hukema R.K., Viville S., Martinat C., Todd P.K., Charlet-Berguerand N. Translation of Expanded CGG Repeats into FMRpolyG Is Pathogenic and May Contribute to Fragile X Tremor Ataxia Syndrome. Neuron. 2017. 93(2): 331–347.
Sellier C., Usdin K., Pastori C., Peschansky V.J., Tassone F., Charlet-Berguerand N. The multiple molecular facets of fragile X-associated tremor/ataxia syndrome. J Neurodev. Disord. 2014. 6(1): 23.
Sharma A., Hoeffer C.A., Takayasu Y., Miyawaki T., McBride S.M., Klann E., Zukin R.S. Dysregulation of mTOR signaling in fragile X syndrome. J. Neurosci. 2010. 30(2): 694–702.
Shelton A.L., Cornish K.M., Godler D., Bui Q.M., Kolbe S., Fielding J. White matter microstructure, cognition, and molecular markers in fragile X premutation females. Neurology. 2017. 88(22): 2080–2088.
Sherman S.L. Premature ovarian failure in the fragile X syndrome. Am. J. Med. Genet. 2000. 97(3): 189–194.
Sterling A., Abbeduto L. Language development in school-age girls with fragile X syndrome. Journal of Intellectual Disability Research. 2012. 56(10): 974–983.
Stöger R., Genereux D.P., Hagerman R.J., Hagerman P.J., Tassone F., Laird C.D. Testing the FMR1 promoter for mosaicism in DNA methylation among CpG sites, strands, and cells in FMR1-expressing males with fragile X syndrome. PLoS One. 2011. 6(8): e23648.
Sullivan A.K., Marcus M., Epstein M.P., Allen E.G., Anido A.E., Paquin J.J., Yadav-Shah M., Sherman S.L. Association of FMR1 repeat size with ovarian dysfunction. Hum. Reprod. 2005. 20: 402–412.
Tassone F., Hagerman R.J., Chamberlain W.D., Hagerman P.J. Transcription of the FMR1 gene in individuals with fragile X syndrome. Am. J. Med. Genet. 2000. 97(3): 195–203.
Tassone F., Beilina A., Carosi C., Albertosi S., Bagni C., Li L., Glover K., Bentley D., Hagerman P.J. Elevated FMR1 mRNA in premutation carriers is due to increased transcription. RNA. 2007. 13(4): 555–562.
Tassone F., Hagerman R.J., Taylor A.K., Hagerman P.J. A majority of fragile X males with methylated, full mutation alleles have significant levels of FMR1 messenger RNA. J. Med. Genet. 2001. 38(7): 453–456.
Tassone F., Iwahashi C., Hagerman P.J. FMR1 RNA within the intranuclear inclusions of Fragile X-Associated Tremor/Ataxia Syndrome (FXTAS). RNA Biology. 2004. 1(2): 103–105.
Tassone F., Iong K.P., Tong T.H., Lo J., Gane L.W., Berry-Kravis E., Nguyen D., Mu L.Y., Laffin J., Bailey D.B., Hagerman R.J. FMR1 CGG allele size and prevalence ascertained through newborn screening in the United States. Genome Med. 2012. 21.4(12): 100. https://doi.org/10.1186/gm401
Todd P.K., Oh S.Y., Krans A., He F., Sellier C., Frazer M., Renoux A.J., Chen K.C., Scaglione K.M., Basrur V., Elenitoba-Johnson K., Vonsattel J.P., Louis E.D., Sutton M.A., Taylor J.P., Mills R.E., Charlet-Berguerand N., Paulson H.L. CGG repeat-associated translation mediates neurodegeneration in fragile X tremor ataxia syndrome. Neuron. 2013. 78(3): 440–455.
Villate O., Ibarluzea N., Maortua H., de la Hoz A.B., Rodriguez-Revenga L., Izquierdo-Álvarez S., Tejada M.I. Effect of AGG Interruptions on FMR1 Maternal Transmissions. Front. Mol. Biosci. 2020. 7: 135.
Welt C.K., Smith P.C., Taylor A.E. Evidence of early ovarian aging in fragile X premutation carriers. J. Clin. Endocrinol. Metab. 2004. 89(9): 4569–4574.
Wheeler A.C., Sideris J., Hagerman R., Berry-Kravis E., Tassone F., Bailey D.B. Jr. Developmental profiles of infants with an FMR1 premutation. J Neurodev Disord. 2016. 8(40). https://doi.org/10.1186/s11689-016-9171-8
Westmark C.J., Sokol D.K., Maloney B., Lahiri D.K. Novel roles of amyloid-beta precursor protein metabolites in fragile X syndrome and autism. Mol. Psychiatry. 2016. 21(10): 1333–1341.
Дополнительные материалы отсутствуют.
Инструменты
Журнал высшей нервной деятельности им. И.П. Павлова