

Журнал физической химии, 2020, T. 94, № 9, стр. 1405-1410
Стационарная фаза на основе сверхсшитого полистирола для капиллярной газовой хроматографии
В. Е. Ширяева a, Т. П. Попова a, А. А. Королев a, А. Ю. Канатьева a, А. А. Курганов a, *
a Российская академия наук, Институт нефтехимического синтеза им. А.В. Топчиева
Москва, Россия
* E-mail: Kurganov@ips.ac.ru
Поступила в редакцию 19.11.2019
После доработки 20.01.2020
Принята к публикации 11.02.2020
Аннотация
Используя микросферические частицы сверхсшитого полистирола, приготовлена капиллярная колонка для газовой хроматографии. Показано, что сверхсшитая стационарная фаза по энтальпии сорбции легких углеводородов примерно соответствует стационарным фазам на основе мембранного полимера поли(триметилсилилпропина), но заметно уступает ему по кинетической эффективности. Отмечено, что прямой синтез в колонке сверхсшитой стационарной фазы на основе сверхсшитого полистирола приводит к получению непористого полимера, не проявляющего разделяющей способности по отношению к легким углеводородам.
Все многообразие стационарных фаз, применяемых в современной газовой хроматографии (ГХ), можно разделить на две большие группы: пористые стационарные фазы и непористые стационарные фазы. Последние часто обозначают как “жидкие” стационарные фазы, поскольку их первые представители были действительно высококипящими органическими жидкостями. В настоящее время большинство стационарных фаз, как первой, так и второй группы – это синтетические высокомолекулярные соединения [1]. Пористые полимерные стационарные фазы также можно разделить на две большие группы: классические макропористые полимеры и полимеры с внутренней пористостью.
Классические макропористые полимеры получают полимеризацией мономеров в присутствии специальных добавок – порогенов, количество и строение которых определяют пористую структуру получаемого полимера [2]. Чтобы стабилизировать пористую структуру макропористых полимеров, полимерные цепи сшивают тем или иным способом, фиксируя их пространственное расположение. Напротив, полимеры с внутренней пористостью не содержат никаких сшивок, а их пористость возникает в связи с невозможностью по стерическим причинам полимерным цепям плотно упаковаться в твердом полимере [3]. Такие полимеры обладают микропористой структурой и повышенным свободным объемом, но с термодинамической точки зрения подобное строение полимерной матрицы не является равновесным, и оно медленно меняется во времени в структуру с более плотной упаковкой полимерных цепей, что, естественно, сопровождается уменьшением свободного объема (так называемое физическое старение полимеров) [4].
Сшитые полимерные структуры более стабильны во времени, но проблема была с получением сшитых микропористых структур, обладающих высокоразвитой внутренней поверхностью и приемлемым объемом пор. Эта проблема была решена с разработкой методов получения, так называемых, сверхсшитых полимеров [5], которые получают сшиванием полимерных цепей в среде хорошего для полимера растворителя. Порогеном в этом случае выступает сам хороший растворитель, что и приводит к получению микропористых структур с высокоразвитой внутренней поверхностью. Свойства сверхсшитых полимеров были прежде всего исследованы в различных сорбционных процессах [5]. В хроматографии исследования проводили на наполненных колонках и также касались главным образом термодинамики сорбции на сверхсшитых сорбентах [6, 7]. Цель данной работы состояла в приготовлении капиллярной колонки типа PLOT со стационарной фазой на основе сверхсшитого полистирола и определении не только термодинамических, но и кинетических характеристик сорбции на сверхсшитых стационарных фазах.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Хроматографические измерения
Все хроматографические измерения выполнены на хроматографе фирмы Шимадзу GC-17A, снабженном ПИД и инжектором с делителем потока. В качестве газа-носителя использовался гелий (класс А). Максимальное входное давление в системе составляло 400 кПa при выходном давлении равном 100 кПа (атмосферное). Тестовые смеси были составлены из алифатических углеводородов С1–С4 и С6–С10. Объем вводимой жидкой пробы составлял 0.1 мкл, температура испарителя 250°C, деление потока газа-носителя 50 : 1. Разделения проводились либо в изотермическом режиме, либо с программированием температуры разделения, как указано в подписях к рисункам. Детектирование проводилось с помощью пламенно-ионизационного детектора при температуре детектора 300°C, потоке водорода 40 мл/мин, потоке воздуха 300 мл/мин и дополнительном потоке гелия (make up) 30 мл/мин. Сбор данных и их первичная обработка проводились с помощью программы ЭкоХром (БойСофт, Россия).
Получение полимеров
Поли(триметилсилилпропин) (ПТМСП) был получен в лаборатории ИНХС РАН под руководством Бермешева М.В. на ниобиевом катализаторе [8] и использовался без дополнительной очистки. Монодисперсный сверхсшитый полистирол в виде сферических частиц диаметром dp = = 5 мкм был получен от проф. Даванкова В.А. (ИНЭОС РАН). Свойства полимера приведены в табл. 1. Низкотемпературная сорбция азота показала наличие в сверхсшитом полистироле также микропор с поверхностью порядка 51 м2/г и объемом 0.024 см3/г со средним размером 0.2 нм.
Приготовление капиллярных колонок
Капиллярная колонка со стационарной фазой на основе поли(триметилсилилпропина) (ПТМСП) была приготовлена методом статического нанесения стационарной фазы на стенки кварцевого капилляра длиной L = 25 м и внутренним диаметром dc = 0.2 мм, как описано в [9]. Толщина пленки стационарной фазы df = 0.25 мкм.
Капиллярная колонка со слоем стационарной фазы на основе сверхсшитого полистирола приготовлена путем заполнения капилляра суспензией полимера в смеси четыреххлористого углерода и ацетона (с добавкой или без добавки “связующего” полимера) и последующим испарением растворителя в динамическом режиме (температура зоны испарения 130°С, температура термостата 90°С). Время пребывания колонки в зоне испарителя – несколько секунд, в термостате – 1 ч. После нанесения стационарной фазы колонку помещали в термостат хроматографа и кондиционировали в токе гелия в течение нескольких часов, до получения стабильной базовой линии и отсутствия “спайков” от незакрепленных частиц сверхсшитого полимера.
Калориметические измерения
Дифференциальная сканирующая калориметрия (ДСК) полимеров проводилась на приборе DSC 3, а термогравиметрический анализ (ТГА) на приборе TGA 2 (оба прибора фирмы Mettler Toledo, Швейцария). ДСК и ТГА выполнены в инертной атмосфере аргона. Интервал температур для ТГА-измерений – от комнатной температуры до 800°С; интервал температур для ДСК-измерений – от 30 до 800°С, скорость нагрева 10 K/мин.
Измерение внутренней поверхности полимеров
Определение удельной поверхности, объема и размера пор проводилось на анализаторе удельной поверхности ASAP-2020МР (Micromeritics, США) Центра коллективного пользования “Новые нефтехимические процессы, полимерные композиты и адгезивы”. Перед измерением образцы кондиционировали при 100°С в вакууме.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Капиллярные колонки с пористым слоем неподвижной фазы (колонки типа PLOT) могут быть приготовлены либо путем синтеза стационарной фазы непосредственно в колонке [10], либо нанесением заранее синтезированной стационарной фазы или ее прекурсора (например, графены) на стенки колонки [11, 12]. Каждый из методов имеет свои преимущества и свои недостатки и a priori трудно предсказать, какой из методов приведет к желаемой цели, поэтому в работе были опробованы оба подхода.
Получение пористого слоя поли(хлорметилстирола) in situ в колонке
Получение сверхсшитых полимеров может быть выполнено различными методами, но наиболее широко оно изучено для реакции Фриделя–Крафтса [5]. Учитывая это обстоятельство, мы выбрали в качестве прекурсора для приготовления колонки полихлорметилстирол, который получили простой радикальной полимеризацией хлорметилстирола. Полимер имел, по данным ГПХ-анализа, молекулярную массу МР = 129 кДа и полидисперсность 1.9.
Нанесенный на стенки колонки динамическим методом слой полихлорметилстирола, как и ожидалось, не показал никакой разделяющей способности по отношению к тестовым смесям сорбатов, поскольку до проведения сшивания полихлорметилстирол в твердом состоянии представляет собой непористый, стеклообразный полимер с высокой температурой стеклования. Пористую структуру стационарная фаза должна была приобрести после ее сшивания, для чего капиллярную колонку заполнили раствором четыреххлористого олова (катализатор реакции Фриделя–Крафтса) в хлористом метилене при комнатной температуре. Однако и после предполагаемой сшивки полимерного слоя, стационарная фаза не показала никакой разделяющей способности по отношению к тестовым смесям. Отсутствие разделяющих свойств однозначно указывает на то, что и после проведения реакции сшивания стационарная фаза сохранила непористую, стеклообразную структуру.
Наиболее вероятная причина неудачной попытки приготовления колонки со слоем стационарной фазы на основе сверхсшитого полимера – это сшивание полимерного слоя в отсутствие растворителя в момент нанесения его на стенки капилляра. Чтобы проверить эту возможность, мы выполнили для полихлорметилстирола термогравиметрический анализ (ТГА) и дифференциальную сканирующую калориметрию (ДСК) и сравнили результаты с аналогичными исследованиями сверхсшитого стирола, полученными Беляковой Л.Д. и др. [7]. На термограмме сверхсшитого полистирола Белякова Л.Д. и др. [7] наблюдали двухступенчатую кривую разложения образца с максимумами при температурах 340°С (потеря 30% массы) и 530°С (потеря оставшихся 70% массы). Эти же две ступени наблюдаются и на дериватограмме поли(хлорметилстирола) (рис. 1), но первая ступень смещена в более высокотемпературную область (370°С), а вторая ступень, наоборот, в сторону более низких температур (450°С). Потеря массы на каждой из ступеней также изменилась: на первой ступени теряется более 50% массы, а на второй не более 30%. Изменение соотношения в потере массы на первой и второй ступенях указывает на то, что процесс термического сшивания полимерных цепей по реакции Фриделя–Крафтса (экзотермический процесс) накладывается на процесс термического разложения полимера (эндотермический процесс). Этот вывод подтверждают данные ДСК полимера (рис. 1), где в области обоих пиков на ДСК кривой наблюдаются лишь очень слабые эндотермические эффекты, которые меняются на слабый экзотермический эффект в конце второго пика. Значительный эндотермический эффект начинает проявляться лишь при температуре выше 600°С, когда происходит карбонизация образца поли(хлорметилстирола).
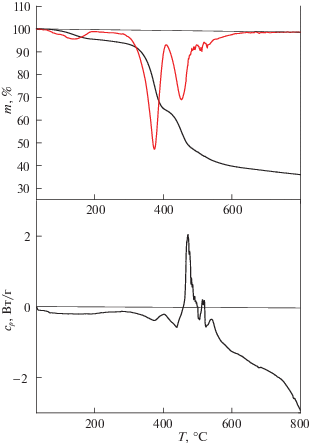
Таким образом, поли(хлорметилстирол) в отсутствие катализаторов реакции Фриделя–Крафтса остается стабильным до ∼300°С и его термическое сшивание происходит лишь при температурах химического разложения. Соответственно, он не мог сшиваться при нанесении стационарной фазы на стенки колонки, если только кварцевые стенки колонки не проявляют каталитического эффекта, что в указанных условиях маловероятно. В то же время, хорошая термостабильность делает этот полимер привлекательным кандидатом для приготовления колонок со стационарной фазой на его основе.
Приготовление колонки на основе заранее синтезированного пористого сверхсшитого полимера
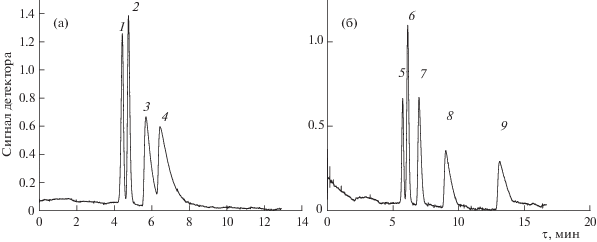
Альтернативный метод получения PLOT капиллярных колонок предполагает закрепление на стенках колонки предварительно синтезированного пористого полимера. Этот метод имеет то преимущество, что пористая структура полимера может быть детально исследована до нанесения стационарной фазы, и она, как правило, предполагается неизменной в процессе приготовления и эксплуатации колонки. Однако нам не удалось найти в литературе каких-либо общих методов закрепления слоя предварительно синтезированного сшитого полимера на стенках колонки, аналогичных динамическому и статическому методам нанесения растворимых полимерных фаз. В этой связи мы попробовали использовать модифицированный динамический метод для закрепления сферических частиц пористого сверхсшитого полимера, используя в качестве “связующего” полисилоксан. Но приготовленная таким методом капиллярная колонка не показала свойств, присущих пористым стационарным фазам, и вела себя как типичная колонка с силоксановой фазой. Вероятно, в процессе нанесения полисилоксан не только “закрепил” сферические частицы сверхсшитого полимера на стенках колонки, но и практически полностью блокировал их поровое пространство. Поэтому следующая попытка была проведена без добавления “связующего”. Была приготовлена суспензия сверхсшитого полимера в равноплотностном растворителе и использован статический метод нанесения стационарной фазы. Этот метод привел к получению капиллярной колонки, способной делить легкие углеводороды (рис. 2), что указывает на микропористый характер стационарной фазы. Селективность колонки при разделении легких углеводородов оказалась заметно ниже той, которая наблюдается у колонки с типично микропористой стационарной фазой на основе поли(триметилсилилпропином) (табл. 2).
Рис. 2.
Разделение углеводородов С1–С4 (а) и С6–С10 (б) на капиллярной колонке (15 м × 0.165 мм) со стационарной фазой на основе сверхсшитого полистирола. Газ-носитель гелий 60 кПа; температурная программа: а – 5 мин при 35°С далее 5 K/мин до 70°С; В – 6 мин 150°С далее 7 K/мин до 200°С. Сорбаты: 1 – метан+этан, 2 – пропан, 3 – изо-бутан, 4 – н-бутан, 5 – н-гексан, 6– н-гептан, 7 – н-октан, 8 – н-нонан, 9 – н-декан.
Более низкая селективность колонки со стационарной фазой на основе сверхсшитого полистирола может быть обусловлена, как его более низкой внутренней поверхностью (см. табл. 1), так и просто меньшим количеством стационарной фазы, закрепленной на стенках капилляра. При нанесении жидких стационарных фаз из растворов предполагается, что в колонке после нанесения остается все количество полимера, изначально содержавшееся в растворе, которым заполняли колонку.
При использовании суспензии полимера, плохо закрепленные частицы полимера выносятся из колонки в процессе нанесения фазы, и, особенно, в процессе кондиционирования колонки. В этой связи какое-то представление о количестве стационарной фазы, оставшейся в колонке, можно получить только из сравнения хроматографических данных по удерживанию стандартных сорбатов (обычно углеводородов). Естественно, что такое сравнение предполагает, что термодинамика сорбции тест-сорбатов на двух фазах будет примерно одинаковой. Чтобы проверить это условие мы определили энтальпию сорбции легких углеводородов на колонке со стационарной фазой на основе сверхсшитого полистирола (рис. 3, табл. 2). Как видно из таблицы, алканы немного более сильно сорбируются на сверхсшитой фазе, чем на ПТМСП, что вероятно связано с наличием ароматических колец в ее структуре. В то же время, наблюдаемое различие явно недостаточно, чтобы вызвать сильное уменьшение в селективности разделения метана и бутана на сверхсшитой фазе. Возможной причиной такого эффекта могут быть стерические затруднения, которые возникают при сорбции на сверхсшитой фазе и которые должны найти свое проявление в кинетических кривых сравниваемых колонок.
Рис. 3.
Зависимости логарифма фактора удерживания сорбатов lnk от обратной температуры колонки 1/Т, K–1. Газ-носитель гелий, 70 кПа, сорбаты: 1 – этан, 2 – пропан, 3 – изо-бутан, 4 – н-бутан.

Кинетические кривые позволяют оценить разделяющую способность стационарной фазы по их способности генерировать теоретические тарелки при изменении условий разделения (длины и диаметра колонок, скорости подвижной фазы и т.д. [13, 14]). Для построения кинетических кривых были измерены зависимости высоты теоретической тарелки H от входного давления газа-носителя на колонке pi (рис. 4), которая аппроксимировались уравнением [9]:
(1)
$\begin{gathered} {{t}_{m}} = {{\left[ {0.75{{C}_{m}}\frac{{p_{i}^{4} - p_{0}^{4}}}{{p_{i}^{3} - p_{0}^{3}}} + {{C}_{S}}} \right]} \mathord{\left/ {\vphantom {{\left[ {0.75{{C}_{m}}\frac{{p_{i}^{4} - p_{0}^{4}}}{{p_{i}^{3} - p_{0}^{3}}} + {{C}_{S}}} \right]} {\left[ {{{N}^{{ - 1}}}\mathop - \limits_{}^{} } \right.}}} \right. \kern-0em} {\left[ {{{N}^{{ - 1}}}\mathop - \limits_{}^{} } \right.}} \\ \left. { - \;2.25B\frac{{\eta (p_{i}^{4} - p_{0}^{4})}}{{{{K}_{V}}{{{(p_{i}^{3} - p_{0}^{3})}}^{2}}}}} \right]. \\ \end{gathered} $Рис. 4.
Оптимальные кинетические зависимости для капиллярных колонок со стационарными фазами на основе ПТМСП и сверхсшитого полистирола; t – время анализа, N – число теоретических тарелок.
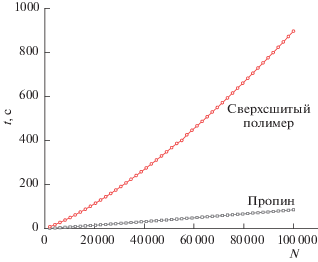
Таким образом, впервые приготовленная капиллярная колонка со стационарной фазой на основе сверхсшитого полистирола показала слабое удерживание и низкую селективность при разделении тестовых сорбатов и плохую кинетическую эффективность, хотя и сравнимую энтальпию сорбции. Слабое удерживание и низкая селективность, скорее всего, объясняются малым количеством стационарной фазы, которое удается закрепить на стенках колонки предложенным методом. Равным образом сверхсшитый полистирол обладал в два раза более низкой внутренней поверхностью, чем ПТМСП. Эти недостатки стационарной фазы на основе сверхсшитого полистирола, вероятно, можно устранить используя более пористый и более мелкодисперсный сверхсшитый полимер, учитывая возрастание склонности к адгезии при уменьшении размера частиц. Одновременно уменьшение размера частиц должно способствовать улучшению кинетики сорбции. Пятимикронные частицы формируют слишком толстый слой стационарной фазы, который будет затруднять кинетику массообмена, как это известно и для толстых слоев жидких стационарных фаз [15].
Работа выполнена при финансовой поддержке Российского фонда фундаментальных исследований (код проекта № 19-03-00040).
Список литературы
Сакодынский К.И., Панина Л.И. Полимерные сорбенты для молекулярной хроматографии. М.: Наука, 1977.
Silverstein M.S., Cameron N.R., Hillmyer M.A. (Eds.) Porous Polymers. NJ: Wiley, Hoboken, 2011.
Yampolskii Yu., Starannikova L., Belov N. et al. // J. Membr. Sci. 2014. V. 453. P. 532.
Morliere N., Vallieres C., Perrin L., Roizard D. // Ibid. 2006. V. 270. P. 123.
Davankov V.A., Tsyurupa M.P. Hypercrosslinked Polymeric Networks and Adsorbing Materials, Synthesis, Structure, Properties and Application. Elsevier, 2010.
Белякова Л.Д., Василевская О.В., Цюрупа М.П., Даванков В.А. // Журн. физ. химии. 1995. Т. 60. С. 696.
Белякова Л.Д., Василевская О.В., Цюрупа М.П., Даванков В.А. // Там же. 1996. Т. 70. С. 1476.
Berezkin V.G., Korolev A.A., Malyukova I.V. et al. // J. Chromatogr. A. 2002. V. 960. P. 151.
Yakubenko E., Korolev A., Chapala P. et al. // Anal. Chim. Acta. 2017. V. 986. P. 153.
Nikolaeva O.A., Patrusheva Y.V., Sidelnikov V.N. // J. Chromatogr. A. 2017. V. 1488. P. 126.
Liu X., Liu X., Li M., Guo L., Yang L. // Ibid. 2013. V. 1277. P. 93.
Kelman S.D. Crosslinking and Stabilization of High Fractional Free Volume Polymers for the Separation of Organic Vapors from Permanent Gases. PhD Tesis. USA: The University of Texas at Austin, 2008. 214 p.
Kurganov A., Kanateva A., Yakubenko E. // J. Sep. Sci. 2016. V. 39. P. 162.
Jespers S., Roeleveld K., Lynen F. et al. // J. Chromatogr. A 2015. V. 1386. P. 81.
Березкин В.Г., Королев А.А., Попова Т.П., Ширяева В.Е. // Журн. аналит. химии. 1989. Т. 44. С. 1402.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии