

Журнал физической химии, 2020, T. 94, № 8, стр. 1215-1221
Расширенное тестирование функционалов плотности для расчета стандартных электродных потенциалов ванадиевых соединений
С. Н. Тусупбаев a, *, Г.М. Кудайбергенова a
a Институт полимерных материалов и технологий
Алматы, Казахстан
* E-mail: samat.tussupbayev@gmail.com
Поступила в редакцию 01.11.2019
После доработки 08.01.2020
Принята к публикации 21.01.2020
Аннотация
Проведено систематическое исследование по выявлению оптимального метода функционала плотности для расчета стандартных электродных потенциалов электрохимического восстановления ванадиевых соединений. Для тестирования выбраны 28 функционалов плотности различного типа. Изучено влияние природы функционала, размера базисного набора, а также учета дисперсионного взаимодействия на точность результатов. Среди использованных в тестировании наименьшее отклонение от экспериментальных данных показано для мета-NGA функционала MN12-L. Предложена оптимальная схема расчета для исследования окислительно-восстановительных реакций ванадиевых соединений.
Системы хранения энергии, основанные на проточных редокс-аккумуляторах (ПРА), привлекли к себе значительный интерес в последнее время [1, 2]. Среди них наиболее изучены ванадиевые проточные редокс-аккумуляторы (ВПРА), впервые предложенные исследовательской группой M. Skyllas-Kazacos [3]. ВПРА обладают такими привлекательными свойствами как высокая электрохимическая активность, емкость, обратимость и низкие эксплуатационные расходы [4].
Имеющиеся на сегодняшний день на рынке ВПРА используют электролиты на основе серной кислоты, содержащие редокс-пару V2+/V3+ в прикатодном пространстве и VO2+/VO$_{2}^{ + }$ – в прианодном. Окислительно-восстановительные реакции, протекающие на аноде и катоде описываются соответственно уравнениями:
(1)
$\begin{gathered} {\text{VO}}_{2}^{ + } + 2{{{\text{H}}}^{ + }} + {{{\text{e}}}^{--}} = {\text{V}}{{{\text{O}}}^{{2 + }}} + {{{\text{H}}}_{2}}{\text{O}}, \\ E^\circ = 1.001\;{\text{В}}, \\ \end{gathered} $(2)
${{{\text{V}}}^{{3 + }}} + {{{\text{e}}}^{--}} = {{{\text{V}}}^{{2 + }}},\quad E^\circ = --0.255\;{\text{В}}.$Экспериментальные величины стандартных потенциалов электродных процессов (1) и (2) взяты из работы [5]. Стандартный потенциал ячейки при концентрациях ионов 1 моль/л и при температуре 25°С, следовательно, составляет 1.256 В.
Гидратированные структуры катионов V2+, V3+, VO2+ и ${\text{VO}}_{2}^{ + }$ и их динамика ранее изучались различными методами. Установлено, что катионы V2+ и V3+ образуют в воде октаэдрические гексааквакомплексы [V(H2O)6]2+ и [V(H2O)6]3+ [6–9]. У катиона VO2+ также октаэдрическая структура с пятью молекулами воды и оксо-лигандом в координационной сфере [9–12]. Октаэдрическая структура [VO(H2O)5]2+ искажена отталкиванием оксо-лиганда и атомов кислорода воды, а осевая связь V–O длиннее средней экваториальной связи V–O. Для гидратированного катиона ${\text{VO}}_{2}^{ + }$ расчеты показали, что бипирамидальная структура [VO2(H2O)3]+ более устойчива [9, 13–15], чем октаэдрическая [VO2(H2O)4]+.
Расчетные методы теории функционала плотности (ТФП) [16] широко используются в последнее время для расчета термодинамических и кинетических параметров, изучения электронного строения и механизмов химических реакций. Это обусловлено их экономичностью с точки зрения использования машинного времени и сравнительно высокой точностью при условии правильного выбора функционала и базисного набора. Применимость метода функционала плотности для расчета стандартного потенциала восстановления пары M3+/M2+, где M = Sc, Ti, V, Cr, Mn, Fe, Co, Ni и Cu, изучалась в работе [17]. Использовался локальный функционал плотности BP86 [18, 19] в сочетании с базисным набором TZVPP [20]. Учет эффектов сольватации проводился по комбинированной схеме (explicit+implicit): до 18 молекул воды в первой и второй сольватной оболочке ионов M3+ и M2+ описывались в явном виде методом функционала плотности, остальной объем растворителя описывался моделью поляризуемого континуума COSMO [21]. Рассчитанная таким методом для пары V3+/V2+ величина стандартного потенциала (–0.17 В) хорошо согласуется с экспериментальным значением (–0.255 В). Одним из недостатком данного подхода является то, что для квантово-механического описания относительно большого числа молекул растворителя требуется учет множества возможных конформаций. Более обоснованно с точки зрения теории в таком случае использование методов молекулярной динамики [22].
В работе [23] для расчета стандартных потенциалов восстановления октаэдрических комплексов [Ti(H2O)6]3+, [V(H2O)6]3+, [Cr(H2O)6]3+, [Mn(H2O)6]3+, [Fe(H2O)6]3+, [Fe(CN)6]3+, [Co(H2O)6]3+, [Co(NH3)6]3+ и [Cu(H2O)6]3+ был использован гибридный функционал B3LYP в сочетании с трехэкспонентным базисным набором. Первоначально учет сольватации проводился по схеме: шесть молекул воды (лигандов) в первой сольватной оболочке ионов, остальной объем растворителя описывался моделью поляризуемого континуума COSMO. Рассчитанные величины стандартных потенциалов оказались систематически завышены на 1.6 В по сравнению с экспериментальными. Поэтому в дальнейшем авторами был использован метод QM/MM, в рамках которого проводилось молекулярно-динамическое моделирование. Ион металла и шесть молекул воды при этом описывались квантово-механически, остальная часть растворителя была представлена 1720 молекулами воды, описываемыми классически с использованием силового поля SPC/E [24]. Это позволило снизить среднеквадратичное отклонение от эксперимента до 0.3 В. Однако следует отметить, что данный подход имеет ограниченную применимость для изучения механизмов окислительно-восстановительных реакций ванадиевых соединений, например, для изучения влияния присадок на устойчивость ванадиевого электролита, реакции полимеризации, саморазряда и пр., вследствие высоких затрат машинного времени. Кроме того, в описанных работах изучалась применимость методов функционала плотности для расчета стандартного электродного потенциала для пары V3+/V2+, но не для других степеней окисления.
В настоящей работе проведено тестирование 28 функционалов плотности, чтобы выбрать недорогой с точки зрения затрат машинного времени метод для аккуратного воспроизведения термодинамических характеристик, а именно стандартных электродных потенциалов окислительно-восстановительных реакций ванадиевых соединений. Для расчетов были выбраны реакции (1) и (2), а также реакция перехода VIV в VIII [5]:
(3)
$\begin{gathered} {\text{V}}{{{\text{O}}}^{{2 + }}} + 2{{{\text{H}}}^{ + }} + {{{\text{e}}}^{--}} = {{{\text{V}}}^{{3 + }}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}}, \\ E_{{\text{3}}}^{ \circ } = 0.337\;{\text{В}}. \\ \end{gathered} $МЕТОДИКА РАСЧЕТА
Все расчеты проводились с использованием програмного комплекса Gaussian 09 [25]. Оптимизация геометрии проводилась с использованием функционала M06-L [26] в сочетании с двухэкспонентным базисным набором def2-SVP [27]. Для полученных геометрий были выполнены расчеты энергии с использованием 28 функционалов плотности различного класса. В качестве локальных были использованы популярные функционалы обобщенного градиентного приближения (generalized gradient approximation (GGA)): BLYP [18, 28], OLYP [28, 29], PBEPBE [30], B97D [31], а также мета-GGA-функционалы TPSS [32], M06-L [26] и M11-L [33]. Помимо GGA- функционалов также тестировались “неделимые” (nonseparable (NGA)) функционалы: N12 [34] и его мета-версия – MN12-L [35]. Особенность последних – в них обменно-корреляционная энергия не может быть представлена в виде суммы обменной и корреляционной составляющих. Тестировались гибридные функционалы с различным вкладом хартри-фоковского обменного взаимодействия: глобально гибридные функционалы O3LYP [28, 29, 36–38], B3LYP*, B3LYP [18, 37–39], B3PW91 [18, 37–41], B971 [42], B972 [43], PBE0 [44], mPW1PW91 [45], а также мета-гибридные функционалы TPSSh*, TPSSh [46, 47], tHCTHhyb [48], M06, M06-2X [49]. Также использовались так называемые пространственно-разделенные (range-separated) гибридные функционалы, в которых вклад хартри-фоковского обменного взаимодействия плавно меняется в зависимости от межэлектронного расстояния: CAM-B3LYP [50], ωB97X-D2 [51], HSEh1PBE [52, 53], N12-SX (NGA) [54], а также мета-гибридные пространственно-разделенные функционалы – M11 [55] и MN12-SX (NGA) [54]. При этом в функционалах HSEh1PBE, N12-SX и MN12-SX точная обменная энергия воспроизводится использованием экранированного кулоновского потенциала (screened Coulomb potential hybrid).
Учет дисперсионного взаимодействия велся методом DFT-D, предложенным Grimme и др. [56] с демпфированием по Becke–Johnson [57] для следующих функционалов: B97-D3(BJ), TPSSh-D3(BJ), B3LYP-D3(BJ), PBE0-D3(BJ).
Тестирование функционалов проводилось с использованием трехэкспонентного базисного набора def2-TZVP [27]. Кроме того, с отдельными функцоналами были использованы базисные наборы def2-SVP, def2-TZVPP и def2-QZVP [27].
Природа стационарных точек была подтверждена аналитическим расчетом колебательных частот в рамках гармонического приближения. Учет сольватации проводился по схеме: шесть молекул воды (лигандов) в первой сольватной оболочке ионов, остальной объем растворителя описывался моделью поляризуемого континуума Solvation Model based on Density (SMD) [58], с улучшенной параметризацией для воспроизведения энергий сольватации. В работе использовалась диэлектрическая постоянная ε = 78.3553 для воды в качестве растворителя.
Стандартные окислительно-восстановительные потенциалы рассчитывались с использованием термохимического цикла Борна–Хабера по формуле:
(4)
${{E}_{{{\text{rel}},{\text{SHE}}}}}({\mathbf{A}}\,{\text{|}}\,{\mathbf{B}}) = - \frac{{{{\Delta }_{{\text{r}}}}G_{{\text{s}}}^{^\circ }({\mathbf{A}}\,{\text{|}}\,{\mathbf{B}})}}{{nF}} - E_{{{\text{abs}}}}^{^\circ }({\text{SHE}}),$(5)
$\begin{gathered} {{\Delta }_{{\text{r}}}}G_{{\text{s}}}^{^\circ }({\mathbf{A}}\,{\text{|}}\,{\mathbf{B}}) = {\text{\;}}{{\Delta }_{{\text{r}}}}G_{{\text{g}}}^{^\circ }({\mathbf{A}}\,{\text{|}}\,{\mathbf{B}}) + \\ + \;{\text{\;}}\mathop \sum \limits_i \,\Delta G_{{{\text{solv}}}}^{^\circ }({{{\mathbf{B}}}_{i}}) - \mathop \sum \limits_i \,\Delta G_{{{\text{solv}}}}^{^\circ }({{{\mathbf{A}}}_{i}}). \\ \end{gathered} $Энергия Гиббса реакции в газовой фазе в свою очередь рассчитывается согласно закону Гесса:
(6)
$\begin{gathered} {{\Delta }_{{\text{r}}}}G_{{\text{g}}}^{^\circ }({\mathbf{A}}\,{\text{|}}\,{\mathbf{B}}) = {\text{\;}}\mathop \sum \limits_i \,{{\Delta }_{{\text{r}}}}G_{{\text{g}}}^{^\circ }({{{\mathbf{B}}}_{i}}) - \\ - \;\mathop \sum \limits_i \,{{\Delta }_{{\text{r}}}}G_{{\text{g}}}^{^\circ }({{{\mathbf{A}}}_{i}}) - G_{{\text{g}}}^{^\circ }({{{\text{e}}}^{ - }}), \\ \end{gathered} $ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Как отмечалось выше, в растворе гидратированный ванадий находится в виде аквакомплексов [6–15], оптимизированные геометрии которых приведены на рис. 1. Уравнения реакций (1)–(3) для гидратированных форм ванадия можно записать следующим образом:
(7)
$\begin{gathered} {{[{\text{V}}{{{\text{O}}}_{2}}{{({{{\text{H}}}_{{\text{2}}}}{\text{O}})}_{3}}]}^{ + }} \cdot ({{{\text{H}}}_{{\text{2}}}}{\text{O}}) + 2{{{\text{H}}}^{ + }} + {{{\text{e}}}^{--}} = \\ = {{[{\text{VO(}}{{{\text{H}}}_{{\text{2}}}}{\text{O}}{{{\text{)}}}_{{\text{5}}}}]}^{{2 + }}},\quad E_{1}^{^\circ }, \\ \end{gathered} $(8)
${{[{\text{V(}}{{{\text{H}}}_{{\text{2}}}}{\text{O}}{{{\text{)}}}_{{\text{6}}}}]}^{{3 + }}} + {{{\text{e}}}^{--}} = {{[{\text{V(}}{{{\text{H}}}_{{\text{2}}}}{\text{O}}{{{\text{)}}}_{{\text{6}}}}]}^{{2 + }}},\quad E_{2}^{^\circ },$(9)
${{[{\text{VO}}{{({{{\text{H}}}_{{\text{2}}}}{\text{O}})}_{{\text{5}}}}]}^{{2 + }}} + 2{{{\text{H}}}^{ + }} + {{{\text{e}}}^{--}} = {{[{\text{V}}{{({{{\text{H}}}_{{\text{2}}}}{\text{O}})}_{{\text{6}}}}]}^{{3 + }}},\quad E_{3}^{^\circ }.$Рис. 1.
Оптимизированные геометрии аквакомплексов [V(H2O)6]2+, [V(H2O)6]3+, [VO(H2O)5]2+ и [VO2(H2O)3]+ · (H2O), рассчитанные методом M06L/def2-SVP.
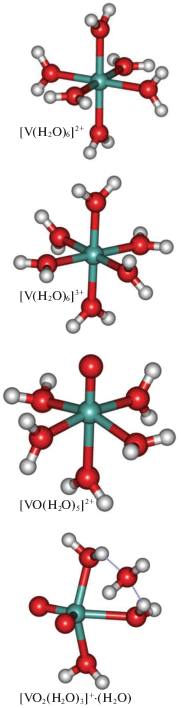
Ионы [V(H2O)6]2+, [V(H2O)6]3+ и [VO(H2O)5]2+ – системы с открытой оболочкой, у которых основными электронными состояниями являются конфигурации с мультиплетностью 4, 3 и 2 соответственно. Согласно расчетам методом M06-L/def2-SVP, для комплексов [V(H2O)6]2+ и [V(H2O)6]3+ низкоспиновые конфигурации с мультиплетностью 2 и 1 выше по энергии высокоспиновых на 32.3 и 39.4 ккал/моль соответственно. Из литературных данных известно, что свободный ион ${\text{VO}}_{2}^{ + }$ в газовой фазе в основном электронном состоянии обладает закрытой электронной оболочкой [61]. Энергетически ближайшее к нему триплетное состояние, различие в энергии составляет 32.1 ккал/моль. Расчеты методом M06-L/def2-SVP показывают, что для гидратированного иона [VO2(H2O)3]+ · (H2O) также характерно основное синглетное состояние, при этом триплетное состояние выше по энергии на 51.0 ккал/моль.
Значения стандартных электродных потенциалов реакций (7)–(9), рассчитанные разными функционалами в сочетании с базисным набором def2-TZVP представлены в табл. 1. Там же приведены максимальное (Δmax) и среднее абсолютное отклонения ($\bar {\Delta }$) рассчитанных стандартных электродных потенциалов реакций от экспериментальных данных. Для удобства функционалы в табл. 1 выстроены по увеличению доли хартри-фоковского обменного взаимодействия (%HF) в общую обменную энергию, причем для пространственно-разделенных функционалов этот вклад указан для коротко- и дальнодействующей компонент обменного функционала. Как видно из этих данных, подавляющее большинство локальных функционалов, т.е. функционалы с нулевым вкладом хартри-фоковского обменного взаимодействия (%HF = 0), значительно занижают стандартные потенциалы $E_{1}^{^\circ }$ и $E_{3}^{^\circ }$. Этим преимущественно обусловлены высокие значения максимального (Δmax) и среднего абсолютного отклонения ($\bar {\Delta }$) для этих функционалов, причем для некоторых из них Δmax > 1 В. Исключение составляют миннесотские функционалы M11-L и M12-NL, предсказывающие потенциалы $E_{1}^{^\circ }$ и $E_{3}^{^\circ }$ очень близкие к экспериментальным. В этом отношении они ведут себя как некоторые гибридные функционалы.
Таблица 1.
Влияние природы функционала плотности на рассчитанные значения стандартных потенциалов реакций (7)–(9), их максимальное (Δmax) и среднее абсолютное отклонения ($\bar {\Delta }$) от экспериментальных данных
| Метод | %HF | $E_{1}^{^\circ }$, В | $E_{2}^{^\circ }$, В | $E_{3}^{^\circ }$, В | Δmax, В | $\bar {\Delta }$, В |
|---|---|---|---|---|---|---|
| BLYP | 0 | –0.183 | –0.104 | –0.646 | 1.184 | 0.773 |
| OLYP | 0 | 0.027 | 0.174 | –0.559 | 0.974 | 0.766 |
| PBEPBE | 0 | –0.177 | –0.082 | –0.652 | 1.178 | 0.780 |
| B97-D3(BJ) | 0 | 0.268 | 0.194 | –0.250 | 0.733 | 0.590 |
| TPSS | 0 | 0.177 | –0.122 | –0.316 | 0.824 | 0.537 |
| M06-L | 0 | 0.406 | 0.283 | –0.122 | 0.595 | 0.531 |
| M11-L | 0 | 0.955 | 0.666 | 0.337 | 0.921 | 0.322 |
| N12 | 0 | 0.000 | –0.161 | –0.599 | 1.001 | 0.677 |
| MN12-L | 0 | 0.962 | –0.170 | 0.234 | 0.103 | 0.075 |
| TPSSh | 10 | 0.577 | 0.084 | 0.028 | 0.424 | 0.357 |
| TPSSh-D3(BJ) | 10 | 0.633 | 0.059 | 0.076 | 0.368 | 0.314 |
| O3LYP | 11.61 | 0.546 | 0.440 | –0.090 | 0.695 | 0.526 |
| TPSSh* | 15 | 0.778 | 0.177 | 0.206 | 0.432 | 0.262 |
| tHCTHhyb | 15 | 0.727 | 0.373 | 0.142 | 0.628 | 0.366 |
| B3LYP* | 15 | 0.506 | 0.282 | –0.028 | 0.537 | 0.466 |
| B3LYP | 20 | 0.800 | 0.421 | 0.232 | 0.676 | 0.327 |
| B3LYP-D3(BJ) | 20 | 0.876 | 0.379 | 0.298 | 0.634 | 0.266 |
| B3PW91 | 20 | 0.860 | 0.419 | 0.265 | 0.674 | 0.296 |
| B971 | 21 | 0.813 | 0.294 | 0.195 | 0.549 | 0.293 |
| B972 | 21 | 0.897 | 0.426 | 0.245 | 0.681 | 0.292 |
| PBE0 | 25 | 0.907 | 0.398 | 0.299 | 0.653 | 0.262 |
| PBE0-D3(BJ) | 25 | 0.950 | 0.380 | 0.336 | 0.635 | 0.229 |
| mPW1PW91 | 25 | 1.035 | 0.465 | 0.417 | 0.720 | 0.278 |
| M06 | 27 | 0.722 | 0.486 | 0.226 | 0.741 | 0.377 |
| BHandHLYP | 50 | 1.942 | 0.656 | 1.295 | 0.958 | 0.937 |
| M06-2X | 54 | 1.721 | 0.670 | 0.982 | 0.925 | 0.763 |
| CAM-B3LYP | 19/65 | 0.985 | 0.489 | 0.355 | 0.744 | 0.260 |
| ωB97X-D | 22.2/100 | 1.201 | 0.375 | 0.499 | 0.630 | 0.331 |
| HSEh1PBE | 25/0 | 0.887 | 0.377 | 0.290 | 0.632 | 0.265 |
| N12-SX | 25/0 | 0.954 | 0.030 | 0.302 | 0.285 | 0.122 |
| MN12-SX | 25/0 | 1.269 | 0.347 | 0.559 | 0.602 | 0.364 |
| M11 | 42.8/100 | 1.565 | 0.342 | 0.694 | 0.597 | 0.506 |
| Эксп. | - | 1.001 | –0.255 | 0.337 | – | – |
Из данных табл. 1 также следует, что в целом гибридные функционалы гораздо точнее локальных в предсказании стандартных потенциалов $E_{1}^{^\circ }$ и $E_{3}^{^\circ }$. В частности, функционалы с вкладом хартри-фоковского обменного взаимодействия 20–25%, например, PBE0, mPW1PW91, B3PW91, B972 и др. дают близкие к экспериментальным значения $E_{1}^{^\circ }$ и $E_{3}^{^\circ }$. При этом они систематически завышают значения потенциала $E_{2}^{^\circ }$, и в результате его значения становятся положительными. Поэтому при сравнительно низком среднем абсолютном отклонении $\bar {\Delta }$, к примеру у функционала PBE0 оно равно 0.262 В, максимальное абсолютное отклонение Δmax при этом достигает 0.653 В.
Гибридные функционалы с меньшим вкладом хартри-фоковского обменного взаимодействия (10–15%), например, TPSSh, TPSSh*, B3LYP* и tHCTHhyb показывают промежуточные результаты между двумя предыдущими группами функционалов.
Многие локальные функционалы правильно воспроизводят знак и дают достаточно близкие к экспериментальным значения $E_{2}^{^\circ }$, например, BLYP (–0.104 В), TPSS (–0.122 В), N12 (–0.161 В) и MN12-L (–0.170 В). В данной работе этого удалось достичь использованием лишь шести молекул воды, рассчитанных методом функционала плотности в явном виде, в отличие от результатов работы [17], в которой потребовалось в явном виде учитывать 18 молекул воды. Гибридные функционалы, с другой стороны, как и в работе [23] систематически завышают значения $E_{2}^{^\circ }$. Причем наибольшие отклонения дают гибридные функционалы с наиболее высоким вкладом хартри-фоковского обменного взаимодействия.
Учет дисперсионного взаимодействия, -D3(BJ), незначительно улучшил точность расчетных методов, для функционалов B3LYP, PBE0, TPPSh значение $\bar {\Delta }$ уменьшилось на 0.061, 0.033 и 0.043 В, соответственно.
Таким образом, наименьшее отклонение от экспериментальных данных получено для мета-NGA-функционала MN12-L со средним и максимальным абсолютным отклонением, равным 0.075 и 0.103 В соответственно. За ним следует пространственно-разделенный гибридный функционал N12-SX с незначительно более высоким $\bar {\Delta }$, равным 0.122 В, но в ∼3 раза большим Δmax (0.285 В).
Далее стандартные потенциалы были рассчитаны с использованием функционалов MN12-L и N12-SX и базисных наборов различного размера. Увеличение размера базисного набора можно продемонстрировать ростом числа базисных функций на примере иона [V(H2O)6]3+: def2-SVP (175), def2-TZVP (303), def2-TZVPP (418), def2-QZVP (786), def2-QZVPP (795). Как видно из табл. 2, для функционала MN12-L наблюдается постепенное снижение величины $\bar {\Delta }$ с увеличением базисного набора. В то же время для функционала N12-SX минимальное отклонение от экспериментальных данных получено с самым компактным базисным набором def2-SVP. Дальнейшее увеличение базисного набора слабо влияет на величину $\bar {\Delta }$, что можно объяснить компенсацией ошибок в случае базисного набора def2-SVP.
Таблица 2.
Влияние размера базисного набора на значения стандартных потенциалов реакций (7)–(9), их максимальное (Δmax) и среднее абсолютное отклонения ($\bar {\Delta }$) от экспериментальных данных
| Метод | $E_{1}^{^\circ }$, В | $E_{2}^{^\circ }$, В | $E_{3}^{^\circ }$, В | Δmax, В | $\bar {\Delta }$, В |
|---|---|---|---|---|---|
| MN12-L | |||||
| def2-SVP | 1.066 | –0.177 | 0.263 | 0.078 | 0.072 |
| def2-TZVP | 0.962 | –0.170 | 0.234 | 0.103 | 0.075 |
| def2-TZVPP | 1.073 | –0.213 | 0.336 | 0.072 | 0.038 |
| def2-QZVP | 1.043 | –0.245 | 0.361 | 0.042 | 0.025 |
| def2-QZVPP | 1.032 | –0.249 | 0.361 | 0.031 | 0.020 |
| N12-SX | |||||
| def2-SVP | 1.046 | –0.113 | 0.294 | 0.142 | 0.077 |
| def2-TZVP | 0.954 | 0.030 | 0.302 | 0.285 | 0.122 |
| def2-TZVPP | 1.033 | –0.006 | 0.391 | 0.249 | 0.112 |
| def2-QZVP | 1.031 | –0.025 | 0.431 | 0.230 | 0.118 |
| Эксп. | 1.001 | –0.255 | 0.337 | – | – |
Таким образом, в результате расширенных тестов для исследования окислительно-восстановительных реакций ванадиевых соединений предлагается следующая схема расчета: оптимизация геометрии с использованием мета-GGA- функционала M06-L и базисного набора def2-SVP; последующий “точечный” расчет мета-NGA-функционалом MN12-L в сочетании с расширенным базисным набором def2-TZVPP. Для дальнейшего увеличения точности результатов можно использовать более дорогой базисный набор def2-QZVPP.
ЗАКЛЮЧЕНИЕ
Таким образом, проведено систематическое исследование по выявлению оптимального метода функционала плотности для расчета стандартных электродных потенциалов электрохимического восстановления ванадиевых соединений. Для тестирования были выбраны 28 функционалов плотности различного типа. Установлено, что большинство локальных функционалов неплохо воспроизводит стандартный электродный потенциал $E_{2}^{^\circ }$ для пары V3+/V2+, однако сильно занижают потенциалы $E_{1}^{^\circ }$ и $E_{3}^{^\circ }$, т.е. пар ${\text{VO}}_{2}^{ + }$/VO2+ и VO2+/V3+ соответственно. Исключение составляют миннесотские функционалы M11-L и M12-NL, предсказывающие потенциалы $E_{1}^{^\circ }$ и $E_{3}^{^\circ }$, близкие к экспериментальным. Популярные гибридные функционалы, например, PBE0, mPW1PW91, B3PW91, B972 и др., с долей хартри-фоковского обменного взаимодействия ∼20–25%, наоборот, достаточно аккуратно предсказывают стандартные электродные потенциалы $E_{1}^{^\circ }$ и $E_{3}^{^\circ }$, но при этом систематически завышают потенциал $E_{2}^{^\circ }$. Схожая картина наблюдается и для пространственно-разделенных гибридных функционалов. Наиболее точным среди них оказался функционал N12-SX, который в меньшей степени завышает потенциал $E_{2}^{^\circ }$ и для которого среднее абсолютное отклонение $\bar {\Delta }$ = 0.122 В.
Непригодными для предсказания стандартных электродных потенциалов электрохимического восстановления ванадиевых соединений оказались функционалы с высоким вкладом хартри-фоковского обменного взаимодействия: BHandHLYP, M06-2X и M11. Учет дисперсионного взаимодействия позволил незначительно снизить значение $\bar {\Delta }$, для функционалов B3LYP, PBE0, TPPSh поправка варьируется в пределах 0.033–0.061 В.
Среди протестированных функционалов наименьшее отклонение от экспериментальных данных дает мета-NGA-функционал MN12-L со средним абсолютным отклонением $\bar {\Delta }$ = 0.075 В. С увеличением базисного набора точность метода можно повысить, снизив $\bar {\Delta }$ до 0.020 В. Предложена оптимальная схема расчета для исследования окислительно-восстановительных реакций ванадиевых соединений.
БЛАГОДАРНОСТИ
Работа выполнена при финансовой поддержке гранта Министерства образования и науки РК (Проект № AP05130375). Для проведения квантово-химических расчетов использовался вычислительный кластер “Национальной научной лаборатории коллективного пользования информационных и космических технологий” при КазНИТУ им. К.И. Сатпаева.
Список литературы
Alotto P., Guarnieri M., Moro F. // Renewable & Sustainable Energy Reviews. 2014. V. 29. P. 325.
Noack J., Roznyatovskaya N., Herr T., Fischer P. // Angew. Chem. Int. Ed. 2015.V. 54. P. 9776.
Skyllas-Kazacos M., Rychcik M., Robins R.G. et al. // J. Electrochem. Soc. 1986. V. 133. P. 1057.
Zhao P., Zhang H., Zhou H. et al. // J. Power Sources. 2006. V. 162. P. 1416.
Bratsch S.G. // J. Phys. Chem. Ref. Data. 1989. V. 18. P. 1.
Akesson R., Pettersson L.G.M., Sandstrom M., Wahlgren U. // J. Am. Chem. Soc. 1994. V. 116. P. 8691.
Benmelouka M., Messaoudi S., Furet E. et al. // J. Phys. Chem. A. 2003. V. 107. P. 4122.
Sepehr F., Paddison S.J. // Chem. Phys. Lett. 2013. V. 585. P. 53.
Larsen S.C. // J. Phys. Chem. A. 2001. V. 105. P. 8333.
Grant C.V., Cope W., Ball J.A. et al. // J. Phys. Chem. B. 1999. V. 103. P. 10627.
Vijayakumar M., Burton S.D., Huang C. et al. // J. Power Sources. 2010. V. 195. P. 7709.
Bühl M., Parrinello M. // Chem.–Eur. J. 2001. V. 7. P. 4487.
Krakowiak J., Lundberg D., Persson I. // Inorg. Chem. 2012. V. 51. P. 9598.
Vijayakumar M., Li L., Graff G. et al. // J. Power Sources. 2011. V. 196. P. 3669.
Jiang Zh., Klyukin K., Alexandrov V. // J. Chem. Phys. 2016. V. 145. P. 114303.
Parr R.G., Yang W. Density Functional Theory of Atoms and Molecules. New York: Oxford University Press, 1989.
Uudsemaa M., Tamm T. // J. Phys. Chem. A. 2003. V. 107. P. 9997.
Becke A.D. // Phys. Rev. A. 1988. V. 38. P. 3098.
Perdew J.P. // Phys. Rev. B. 1986. V. 33. P. 8822.
Schäfer A., Huber C., Ahlrichs R.J. // Chem. Phys. 1994. V. 100. P. 5829.
Klamt A., Schüürmann G.J. // Chem. Soc. Perkin Trans. 2. 1993. P. 799.
Ryu H., Park J., Kim H.K., Park et al. // Organometallics. 2018. V. 37. P. 3228.
Wang L.P., Van Voorhis T. // J. Chem. Theory Comput. 2012. V. 8. P. 610.
Berendsen H.J.C., Grigera J.R., Straatsma T.P. // J. Phys. Chem. 1987. V. 91. P. 6269.
Frisch M.J., Trucks G.W., Schlegel H.B. et al. Gaussian 09 (Gaussian, Inc., Wallingford CT, 2009).
Zhao Y., Truhlar D.G. // J. Chem. Phys. 2006. V. 125. P. 1.
Weigend F., Ahlrichs R. // Phys. Chem. Chem. Phys. 2005. V. 7. P. 3297.
Lee C., Yang W., Parr R.G. // Phys. Rev. B. 1988. V. 37. P. 785.
Handy N.C., Cohen A.J. // Mol. Phys. 2001. V. 99. P. 403.
Perdew J.P., Burke K., Ernzerhof M. // Phys. Rev. Lett. 1996. V. 77. P. 3865.
Grimme S. // J. Comp. Chem. 2006. V. 27. P. 1787.
Tao J.M., Perdew J.P., Staroverov V.N. et al. // Phys. Rev. Lett. 2003. V. 91. P. 146401.
Peverati R., Truhlar D.G. // J. Phys. Chem. Lett. 2012. V. 3. P. 117.
Peverati R., Truhlar D.G. // J. Chem. Theory and Comput. 2012. V. 8. P. 2310.
Peverati R., Truhlar D.G. // Phys. Chem. Chem. Phys. 2012. V. 10. P. 13171.
Cohen A.J., Handy N.C. // Mol. Phys. 2001. V. 99. P. 607.
Slater J.C. Quantum Theory of Molecules and Solids. V. 4. The Self-Consistent Field for Molecules and Solids. New York: McGraw-Hill, 1974. 640 p.
Vosko S.H., WIlk L., Nusair M. // Can. J. Phys. 1980. V. 58. P. 1200.
Becke A.D. // J. Chem. Phys. 1993. V. 98. P. 5648.
Perdew J.P., Chevary J.A., Vosko S.H. et al. // Phys. Rev. B. 1992. V. 46. P. 6671.
Perdew J.P., Wang Y. // Phys. Rev. B. 1992. V. 45. P. 13244.
Hamprecht F.A., Cohen A., Tozer D.J. et al. // J. Chem. Phys. 1998. V. 109. P. 6264.
Wilson P.J., Bradley T.J., Tozer D.J. // Ibid. 2001. V. 115 P. 9233.
Adamo C., Barone V. // Ibid. 1999. V. 110. P. 6158.
Adamo C., Barone V. // Ibid. 1998. V. 108 P. 664.
Tao J.M., Perdew J.P., Staroverov V.N. et al. // Phys. Rev. Lett. 2003. V. 91. P. 146401.
Staroverov V.N., Scuseria G.E., Tao J. et al. // J. Chem. Phys. 2003. V. 119. P. 12129.
Boese A.D., Handy N.C. // Ibid. 2002. V. 116. P. 9559.
Zhao Y., Truhlar D.G. // Theor. Chem. Acc. 2008. V. 120. P. 215.
Yanai T., Tew D., Handy N. // Chem. Phys. Lett. 2004. V. 393. P. 51.
Chai J.-D., Head-Gordon M. // Phys. Chem. Chem. Phys. 2008. V. 10. P. 6615.
Krukau A.V., Vydrov O.A., Izmaylov A.F. // J. Chem. Phys. 2006. V. 125. P. 224106.
Henderson T.M., Janesko B.G., Scuseria G.E. //Ibid. 2008. V. 128. P. 194105.
Peverati R., Truhlar D.G. // Phys. Chem. Chem. Phys. 2012. V. 14. P. 16187.
Peverati R., Truhlar D.G. // J. Phys. Chem. Lett. 2011. V. 2. P. 2810.
Grimme S., Antony J., Ehrlich S. et al. // J. Chem. Phys. 2010. V. 132. P. 154104.
Grimme S., Ehrlich S., Goerigk L. // J. Comp. Chem. 2011. V. 32. P. 1456.
Marenich A.V., Cramer C.J., Truhlar D.G. // J. Phys. Chem. B. 2009. V. 113. P. 6378.
Kelly C.P., Cramer C.J., Truhlar D.G. // J. Phys. Chem. B. 2006. V. 110. P. 16066.
Kelly C.P., Cramer C.J., Truhlar D.G. // J. Phys. Chem. B. 2007. V. 111. P. 408.
Koyanagi G.K., Bohme D.K., Kretzschmar I. et al. // J. Phys. Chem. A. 2001. V. 105. P. 4259.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии