

Цитология, 2022, T. 64, № 6, стр. 544-556
Тубацин, ингибитор гистондеацетилазы 6, вызывает ацетилирование α-тубулина, блок клеточного цикла, старение и подавление миграции фибробластов мыши, трансформированных онкогенами E1A и cHa-ras
А. Н. Кукушкин 1, *, С. Б. Светликова 1, Н. Д. Аксенов 1, В. А. Поспелов 1
1 Институт цитологии РАН
194064 Санкт-Петербург, Россия
* E-mail: kan@incras.ru
Поступила в редакцию 28.05.2022
После доработки 08.07.2022
Принята к публикации 20.07.2022
- EDN: OJFNBN
- DOI: 10.31857/S0041377122060062
Аннотация
Ингибиторы гистондеацетилазной активности широко используются для остановки пролиферации раковых клеток в клинических испытаниях in vivo и в исследованиях на линиях опухолевых клеток in vitro. Кроме подавления пролиферации опухолевых клеток эти ингибиторы способны индуцировать либо клеточное старение, либо апоптотическую гибель клеток и аутофагию. Гистондеацетилаза 6 (HDAC6, класс II) отличается от других HDAC (класса I) преимущественно цитоплазматической локализацией и низкой деацетилазной активностью в отношении гистонов. Более того, HDAC6 деацетилирует ряд негистоновых белков, включая α-тубулин – компонент микротрубочек, тем самым влияя на их стабильность. Оверэкспрессия HDAC6 была обнаружена в различных линиях опухолевых клеток и индуцированных опухолях мышей. Имеющиеся данные свидетельствуют о том, что HDAC6 участвует в контроле процесса аутофагии, поскольку ацетилирование α-тубулина необходимо для слияния аутофагосом с лизосомами. В настоящей работе мы сравнивали эффекты, производимые ингибитором HDAC бутиратом натрия и тубацином, который является специфическим ингибитором HDAC6, на E1A + Ras-трансформированные фибробласты мыши (линия mERas). Бутират натрия ингибирует активность HDAC класса I (но не HDAC6). Оказалось, что тубацин вызывает те же эффекты, что и бутират натрия, в отношении подавления роста клеток, индукции блока клеточного цикла на границе фаз G1/S и клеточного старения. Поскольку тубацин индуцирует ацетилирование α-тубулина, можно предположить, что определенный уровень ацетилирования α-тубулина необходим для пролиферации, старения и миграции клеток mERas.
Ингибиторы HDAC широко используются для подавления пролиферации опухолевых клеток, так как вызывают блок клеточного цикла, клеточное старение или апоптотическую гибель клеток (Shao et al., 2004; Newbold et al., 2016). Предполагается, что эти эффекты, по крайней мере частично, опосредованы ацетилированием нуклеосомных гистонов, что в свою очередь приводит к активации генов-супрессоров опухолей и ингибированию экспрессии генов, специфичных для пролиферации. Важно отметить, что большинство из этих процессов может быть реализовано в опухолевых клетках с мутантным белком р53, часто стабилизированным в опухолях человека. Кроме того, ингибиторы HDAC вызывают необратимую остановку клеточного цикла в опухолевых клетках, экспрессирующих дикий тип p53, за которой следует либо клеточное старение, либо апоптотическая гибель клеток. Стареющие клетки не делятся, но все еще сохраняют жизнеспособность и секретируют различные факторы роста и цитокины, поддерживающие так называемый секреторный фенотип (SASP) (Rodier, Campisi, 2011; Laberge et al., 2015). Тем не менее, такое покоящееся состояние in vivo потенциально опасно как для этих, так и для окружающих клеток. Стареющие клетки могут перезапустить пролиферацию при определенных условиях, в то время как секретируемые ими факторы могут индуцировать трансформацию окружающих нормальных клеток. Следовательно, в практических клинических целях представляется перспективным индуцировать апоптотическую гибель стареющих опухолевых клеток, но это не всегда возможно из-за активации антиапоптотических факторов семейства Bcl-2 (Adams, Cory, 2007; Delbridge, Strasser, 2015).
Ингибиторы HDAC также могут препятствовать процессу аутофагии, которая играет важную защитную функцию в опухолевых клетках. Это особенно важно в тех случаях, когда эти ингибиторы вызывают старение и гипертрофию клеток. Необходимо своевременно элиминировать стареющие клетки, которые продуцируют избыточное количество белков и других компонентов (Rodier, Campisi, 2011). Переход клетки от цитопротекторной аутофагии к ее цитотоксической форме требует тщательного изучения механизмов аутофагии в опухолевых клетках (Mathew et al., 2007). Следовательно, использование ингибиторов HDAC, которые нацелены на цитоплазматические деацетилазы (Verdel et al., 2000), может обеспечить новый подход для подавления пролиферации опухолевых клеток и их сопутствующей элиминации.
В представленной работе мы использовали фибробласты мыши, трансформированные онкогенами E1A и cHa-Ras (линия mERas), для сравнения эффектов, продуцируемых ингибитором HDAC бутиратом натрия, который ингибирует активность HDAC класса I (но не активность цитоплазматической HDAC6) и тубацином, который является специфическим ингибитором HDAC6 и способствует ацетилированию α-тубулина. Наши данные показывают, что тубацин способен запускать большинство клеточных событий, вызываемых ингибиторами HDAC класса I, а именно бутиратом натрия: G1/S-блок клеточного цикла, клеточное старение и подавление миграции клеток. Поскольку тубацин преимущественно индуцирует ацетилирование α-тубулина, можно предположить, что высокий уровень ацетилирования α-тубулина в микротрубочках необходим для пролиферации, старения и клеточной миграции фибробластов мыши, трансформированных онкогенами E1A и Ras.
МАТЕРИАЛ И МЕТОДИКА
Клетки и реактивы. Эмбриональные фибробласты мыши (MEF) выделяли из эмбрионов через 14–15 сут после начала беременности. Стабильные трансформированные клетки MEF (линия mERas) получали с помощью кальций-фосфатной трансфекции онкогенов E1A аденовируса Ad5 и активированного cH-a-Ras (Поспелова и др., 1990). Клетки mERas выращивали на среде DMEM, содержащей 10% эмбриональной бычьей сыворотки (FCS; HyClone GE, США). Растворенный в ДМСО тубацин (Sigma, США) и водный раствор бутирата натрия (Sigma) использовали в концентрациях 2.5–15 мкM и 4 мM соответственно. Растворенный в ДМСО рапамицин (Calbiochem, США) использовали в конечной концентрации 500 нM.
Для оценки пролиферативной активности клетки mERas высевали в чашки диаметром 60 мм. Число клеток считали на стеклах KOVA (Hycor Biomed, Великобритания). Когда число клеток достигало 35 × × 104, их обрабатывали 10 мкМ тубацина в течение 24, 48 или 72 ч. Необработанные клетки служили контролем. Кривые роста строили, исходя из четырех независимых экспериментов.
Жизнеспособность клеток mERas оценивали с помощью набора Count and Viability (Millipore, США) на цитометре Muse Cell Analyzer (Millipore) по методике, рекомендованной производителем.
Анализ распределения клеток по фазам клеточного цикла. Использовали проточную цитометрию, как описано ранее (Nelyudova et al., 2007). Клетки промывали фосфатно-солевым буферным раствором (PBS) и пермеабилизовали 30 мин 0.01%-ным сапонином. Затем клетки инкубировали в PBS, содержвщем 40 мкг/мл йодистого пропидия, 0.1 мг/мл РНКазы A (все от Sigma) 15 мин при 37°С и анализировали на цитометре CytoFLEX (Beckman, США). Популяции контрольных и обработанных клеток в разных фазах цикла сравнивали с помощью цитометрического светорассеяния и анализировали в программе Win MDI, v.2.8. Количество апоптотических клеток оценивали по относительной площади субдиплоидного пика на гистограммах проточной цитометрии. Для подсчета апоптотических клеток в двумерном распределении использовали набор Аннексин V-FITC (Abcam, США) в сочетании с йодистым пропидием.
МТТ-тест на жизнеспособность клеток. Жизнеспособность клеток оценивали посредством колориметрического анализа с помощью 3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолийбромидом (MTT, Sigma). Количество образовавшегося формазанового продукта коррелирует с числом живых клеток. Приблизительно по 10 тыс. клеток сеяли в 96-луночные планшеты и обрабатывали в течение 1 сут 2.5 мкМ тубацина и (или) 4 мМ бутиратом натрия в среде ДМЕМ. Далее в лунки добавляли МТТ (0.5 мг/мл), растворенный в PBS, и клетки культивировали в течение 1.5 ч при 37°С в CO2-инкубаторе. Полученные осадки формазана растворяли в ДМСО (Sigma). Поглощение измеряли при длине волны 570 нм на планшетном спектрофотометре Multiskan-EX (Thermo Sci., США). Каждый эксперимент повторяли шесть раз с последующим вычислением средних значений и их стандартных ошибок.
Иммуноблотинг. Клеточные экстракты получали, лизируя клетки в буфере RIPA, содержащем раствор PBS, 1% Igepal CA-630, 0.5% дезоксихолата натрия, 0.1% SDS, 1 мМ DTT, набор ингибиторов протеаз (пепстатин А, леупептин, апротинин (все в концентрации 1 мкг/мл), 1 мМ PMSF) и фосфатаз (1 мМ ортованадат натрия, 2.5 мМ EGTA, 20 мМ β-глицерофосфат натрия, 10 мМ фторид натрия) (все упомянутые здесь вещества получены от Sigma). Концентрацию белка в клеточных экстрактах определяли методом Брэдфорд (Bradford, 1976). Пробы по 50 мкг белковых экстрактов разделяли с помощью электрофореза в 10–15%-ном ПААГ, переносили на мембрану PVDF Immobilon-P (Millipore, США) с размером пор 0.45 мкм. Эти мембраны инкубировали с первичными антителами в буфере TBS с 5%-ным BSA в соответствии с рекомендациями производителя. Маркерами молекулярной массы служил набор окрашенных стандартных белков SeeBluePlus2 (Invitrogen, США). В качестве вторых антител использовали козьи антитела против иммуноглобулинов кролика или кроличьи антитела против иммуноглобулинов мыши (все от Sigma), конъюгированные с пероксидазой хрена. Белки на мембранах визуализировали с помощью наборов ECL Optiblot (Abcam) или Super Signal West Femto Maximum Sensitivity Substrate (Thermo Sci.), используя систему гель-документации PXi6 Access (Syngene, Великобритания). Нужные белковые полосы денситометрировали и нормализовали относительно контрольных полос и стандартного белка, обычно α-тубулина. Иммуноблотинг определенных белков повторяли не менее 3 раз. На рисунках приведены иммуноблоты, в которых интенсивности интересующих белковых полос близки средним значениям полос, полученных в нескольких электрофорезах. Погрешности определения интенсивности белковых полос обычно составляли ±0.1 отн. ед.
Использовали первичные антитела к следующим белкам: α-тубулину (T5168; Sigma), LC3 (PM036; MBL, США), ацетилированному по лизину-40 α-тубулину (cs-5335), ацетилированному по лизинам-9/14 гистону H3 (cs-9677), фосфо-S6 (cs-2211), S6 (cs-2317), фосфо-4E-BP1 (cs-9459), SQSTM1/p62 (cs-5114), фосфо-ULK1 по Ser555 (cs-5869) и Ser757 (cs-14202), фосфо-AMPKα по Thr172 (cs-2535) (все от Cell Signaling, США).
Миграция клеток в монослое. Этот анализ проводили, как описано ранее (Romanov et al., 2010). Клетки mERas росли на чашках 35 мм 1 сут, затем в монослое делали рану путем удаления клеток с помощью наконечника 200 мкл. Оставшиеся прикрепленные клетки дополнительно промывали средой DMEM. Для оценки влияния тубацина или бутирата натрия на миграцию клеток в рану, их культивировали в среде DMEM дополнительно в течение 48 ч в присутствии тубацина и (или) бутирата, или без этих ингибиторов (контроль). Затем раны фотографировали под микроскопом Axiovert с 5-кратной линзой. Интегральную плотность клеток считали на 5 равных участках для каждой раны с использованием программы ImageJ. Достоверность различий оценивали с помощью t-критерия Стьюдента.
Протеасомная активность. Химотрипсин-, трипсин- и каспаза-подобные пептидазные активности протеасом определяли, используя флуорогенные субстраты Succ-LLVY-AMC, Ac-RLR-AMC и Z-LLE-AMC соответственно в концентрации 50 мкМ, которые растворяли в буфере, содержащем 50 мМ трис-HCl, pH 7.5, 1 мМ MgCl2, 10 мМ KCl, 1 мМ ДТТ, 5 мМ АТФ (Sigma). Клеточные экстракты инкубировали с субстратами 45 мин при 37°С, как было описано ранее (Kulichkova et al., 2010). Количество высвобожденного вещества АМС определяли по интенсивности флуоресценции с помощью флуориметра Glomax Multi (Promega, США), используя длину волны возбуждения и эмиссии соответственно 365 и 440 нм. Для контроля специфичности перед добавлением субстратов клеточные экстракты обрабатывали протеасомным ингибитором MG132 (100 мкМ; Millipore) или его растворителем для контроля (ДМСО) в течение 20 мин при 4°С.
Анализ клеточного старения. Старение клеток контролировали путем анализа активности β-галактозидазы (SA-β-gal), связанной со старением, как описано ранее (Dimri et al., 1995). Клетки mERas выращивали на покровных стеклах 72 ч. Окрашивание SA-β-gal после обработки фиксированных клеток раствором X-gal (Sigma) наблюдали в проходящем свете и фотографировали под микроскопом Pascal LSM5 (Zeiss, Германия) при увеличении объектива 40×.
Анализ аутофагии. Уровень аутофагии в клетках оценивали, используя набор CYTO-ID (Enzo Life Sci., США), который содержал флуоресцентный краситель, специфически и количественно связывающийся с аутофагическими вакуолями. Живые суспендированные клетки mERas инкубировали с красителем CYTO-ID в течение 30 мин при 37°C в темноте в соответствии с протоколом производителя и анализировали с помощью проточной цитометрии при длине волны 488 нм. Эти эксперименты повторяли не менее 4 раз.
Статистический анализ. Представленные здесь средние значения количественных данных и их средние квадратичные отклонения являются результатами по крайней мере 3-х независимых экспериментов.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Тубацин индуцирует блок клеточного цикла на границе G1 и S фаз. Ранее мы показали (Abramova et al., 2006; Romanov et al., 2010), что бутират натрия (ингибитор HDAC класса I) способен индуцировать как необратимый блок клеточного цикла, так и клеточное старение в клетках mERas. В данной работе мы хотели проверить, может ли специфический ингибитор тубацин цитоплазматической HDAC6 класса II оказывать такое же действие на клетки mERas, как и бутират натрия. Известно, что бутират не ингибирует активность HDAC6 (Guardiola, Yao, 2002; Davie, 2003) и способен индуцировать ацетилирование α-тубулина, хотя и в меньшей степени по сравнению с тубацином (Zhang et al., 2003). Данные, представленные на рис. 1а, показывают, что тубацин значительно увеличивает ацетилирование α-тубулина по сравнению с бутиратом натрия, что хорошо видно при использовании оптимальной экспозиции ECL. Это различие в активностях двух ингибиторов HDAC для субстрата α-тубулина оценивается в десятки раз, причем оно проявляется при минимальной используемой концентрации тубацина – 2.5 мкМ (рис. 1а). Такая концентрация была предложена ранее (Haggarty et al., 2003) как действенная в клетках A549. Влияние бутирата натрия на ацетилирование α-тубулина очень низкое по сравнению с тубацином и может наблюдаться только после длительной экспозиции мембраны в процессе ECL (данные не показаны). Поэтому при их совместном действии вклад 4 мМ бутирата натрия мал и практически теряется на фоне действия 2.5 мкМ тубацина (рис. 1а). С другой стороны, тубацин на порядок меньше влияет на ацетилирование таких неспецифических для него субстратов, как гистоны, в частности гистон H3 по сравнению с бутиратом натрия (рис. 1б), что согласуется с другими исследованиями (Haggarty et al., 2003; Depetter et al., 2019).
Рис. 1.
Тубацин (Туб) способствует накоплению ацетилированного α-тубулина в цитоплазме клеток mERas. а – Имуноблот с антителами к ацетилированной (Ац) и неацетилированной формам α-тубулина после электрофореза клеточных экстрактов (50 мкг) в 10%-ном ПААГ; клетки обрабатывали 2.5 мкМ Туб и (или) 4 мМ бутирата натрия (Бут) в течение 24 или 72 ч; нормализованные значения (отношения интенсивности полос ацетилированной и неацетилированной форм α-тубулина) показаны между панелями. б – Иммуноблот с антителами к гистону H3, ацетилированному по лизинам 9/14, и к α-тубулину после электрофореза в 12.5%-ном ПААГ белков (50 мкг) из клеток mERas, обработанных 5 мкМ Туб или 4 мМ Бут в течение 24 или 72 ч; К – контрольные необработанные клетки. Положение маркерного белка 16 кД указано стрелкой справа; нормализованные значения (отношения интенсивности полос ацетилированного Н3 к α-тубулину) показаны между панелями.

Кроме того, тубацин оказывает сильное антипролиферативное действие, о чем свидетельствуют данные проточной цитометрии и анализа жизнеспособности клеток (МТТ) (рис. 2а, б; рис. 3а). Согласно данным проточной цитометрии (рис. 2а), тубацин способен индуцировать остановку клеточного цикла перед фазой S и уменьшать долю вовлеченных в S‑фазу клеток. Следует отметить, что тубацин несколько менее эффективен в индукции G1/S-блока клеточного цикла, чем бутират натрия в определенных экспериментальных условиях. Как показано другими авторами (Namdar et al., 2010), в трансформированных клетках LNCaP тубацин также может индуцировать остановку в фазе G1, и, судя по анализу транскриптов, он уменьшает экспрессию 72 генов, из которых 40% относятся к клеточному циклу, включая специфические гены для перехода G1/S. Кроме того, тубацин способен ингибировать пролиферацию других опухолевых клеток (линий PC3 клеток простаты и TNBC молочной железы) и повышать экспрессию гена p21Waf1 (Sobue et al., 2016; Yu et al., 2017).
Рис. 2.
Длительное воздействие тубацина (Туб) вызывает G1/S-блок клеточного цикла и гипертрофию клеток mERas, но не их апоптотическую гибель. а – Распределение клеток по фазам клеточного цикла в контроле и после действия 5 мкМ Туб или 4 мМ бутирата натрия (Бут) в течение 24, 48 и 72 ч по данным проточной цитометрии. На столбцах указаны средние значения (%) доли клеток в каждой фазе и их погрешности (вертикальные отрезки) из 5 экспериментов. б – Распределение клеток по фазам клеточного цикла по данным проточной цитометрии контрольных клеток mERas (К) и клеток после действия 5 мкМ тубацина (Туб) в течение 72 ч; процентное содержание апоптотических клеток оценивали по площади субдиплоидных пиков ДНК, которые обозначены стрелками. в – Размеры клеток относительно контрольных клеток (первый столбец), определенные по светорассеиванию при проточной цитометрии; указаны средние размеры и их ошибки из 5 экспериментов.

Рис. 3.
Тубацин снижает жизнеспособность клеток mERas и замедляет их пролиферацию. а – МТТ-анализ клеток mERas, культивируемых с 2.5 мкМ тубацина (Туб), или с 4 мМ бутирата натрия (Бут), или с двумя реагентами вместе (Туб + Бут) в течение 24 ч; К – контрольные клетки; показаны средние количества живых клеток относительно контроля (К) и их ошибки из 4 экспериментов. б – Кривые роста контрольных клеток mERas (К) и клеток, обработанных 10 мкМ тубацина (Туб) в течение 24, 48 и 72 ч.
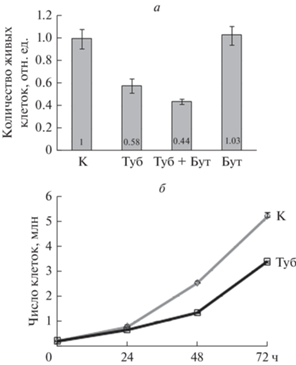
Тем не менее, сам по себе тубацин не вызывал апоптотической гибели клеток mERas в пределах используемых концентраций (2.5–10 мкМ), что следует из малой величины субдиплоидного пика ДНК на рис. 2б. Однако в отличие от клеток mERas, в других типах опухолевых клеток, например, в клетках острого лимфобластного лейкоза, тубацин может индуцировать апоптоз наряду с подавлением пролиферации (Aldana-Masangkay et al., 2011). Параллельно с индукцией G1/S-блока клеточного цикла тубацин снижает их жизнеспособность, которую оценивали по МТТ-тесту (рис. 3а) и также уменьшает количество растущих клеток, судя по анализу кривых роста (рис. 3б). Таким образом, тубацин снижает жизнеспособность клеток mERas в большей степени, чем бутират натрия.
Тубацин способствует гипертрофии и старению трансформированных клеток mERas. Измерение прямого светорассеяния методом проточной цитометрии клеток mERas, обработанных тубацином, показало, что тубацин увеличивает размер клеток (вызывает их гипертрофию) (рис. 2в), что считается необходимым шагом для развития клеточного старения. Хотя гипертрофия клеток выражена не в такой степени, как в случае бутирата натрия, тем не менее, длительная обработка тубацином в течение 72 ч вызывает экспрессию маркера стареющих клеток SA-β-Gal (окрашивание показано на рис. 4а). В совокупности данные свидетельствуют о том, что ацетилирование α-тубулина в трансформированных клетках mERas после действия тубацина способствует остановке клеточного цикла в фазе G1 с последующей индукцией гипертрофии и старения клеток. Таким образом, ацетилирование гистонов, индуцированное обработкой клеток бутиратом натрия и сопровождающееся индукцией бутират-опосредованного клеточного старения (Romanov et al., 2010), не является единственным условием для осуществления старения.
Рис. 4.
Длительная обработка клеток mERas тубацином вызывает клеточное старение в той же степени, что и бутират натрия, а также препятствует миграции клеток в рану. а – Клетки mERas выращивали в течение 72 ч на покровных стеклах в среде DMEM (контроль) или в присутствии 5 мкМ тубацина (Туб) или 4 мМ бутирата натрия (Бут), затем их окрашивали на SA-β-gal; масштабный отрезок под фотографиями справа – 25 мкм. б – Зарастание раны на монослое клеток, нанесенной через 24 ч культивирования, в среде DMEM (контроль зарастания), в присутствии 2.5 мкМ тубацина (Туб), или (и) 4 мМ бутирата натрия (Бут) в течение еще 48 ч культивирования; средние значения интегрированных плотностей клеток в ране показаны под фотографиями. За 1 ед. отн. плотности принимали плотность в местах раны, где отсутствуют клетки.

Тубацин ингибирует миграцию клеток в монослое. Ранее мы показали, что после обработки бутиратом натрия клеток mERas значительно снижается миграция, и эта особенность сильно зависит от экспрессии гена p21Waf1 (Romanov et al., 2010). Для оценки подвижности клеток широко используется процедура миграции клеток в рану in vitro (зарастание раны в монослое). Результаты, представленные в рис. 4б, показывают, что тубацин также ингибировал миграцию клеток mERas в рану после обработки в течение 48 ч, хотя и в меньшей степени, чем бутират натрия. Для сравнения эффектов, производимых бутиратом и тубацином, с помощью программы ImageJ считали интегральные плотности клеток, мигрировавших в рану, на 5 равных полях соответствующих ран монослоя.
Различия между заросшей раной контрольных клеток (в среде DMEM) и ранами, культивируемыми в присутствии тубацина и (или) бутирата натрия (рис. 4б) являются значимыми при P = 0.005 в соответствии с t-критерием Стьюдента. Можно предположить, что индуцированное тубацином ацетилирование α-тубулина служит важным фактором уменьшения стремлений клеток к миграции и метастазированию. В другой работе (Ding et al., 2014) также показано, что тубацин значительно подавлял миграцию и инвазию клеток лимфомы Беркитта. Таким образом, ингибирование HDAC6 тубацином и сопутствующее гиперацетилирование α-тубулина способствуют таким изменениям структуры цитоскелета, которые снижают миграционный потенциал трансформированных клеток.
Активность mTOR и аутофагия. Выше было показано, что ингибирование активности HDAC6 подавляет рост и миграцию клеток mERas, а также индуцирует старение клеток. HDAC6 имеет цитоплазматическую локализацию и не участвует в деацетилировании гистонов, тогда как она деацетилирует α-тубулин – компонент микротрубочек, тем самым влияя на их стабильность (Verdel et al., 2000; Matsuyama et al., 2002; Asthana et al., 2013). Следовательно, ингибирование HDAC6, способствующее ацетилированию α-тубулина, может мешать другому сигнальному пути и кооперировать с противораковыми препаратами, вызывающими деполимеризацию микротрубочек. Например, в клетках немелкоклеточного рака легкого активность HDAC6 поддерживает высокий уровень экспресии рецептора Notch1 и соответствующий сигнальный путь, важный для выживания и пролиферации этих опухолевых клеток, поэтому тубацин и соединения, подобные ингибиторам HDAC, могут быть потенциальными терапевтическими агентам для пациентов с таким раком (Deskin et al., 2020). Тубацин в низких концентрациях способствует пролиферации стволовых стромальных клеток костного мозга и усиливает их механические свойства при увеличении содержания ацетилированного α-тубулина, причем его действие опосредуется через MEK/ERK-сигнальный каскад (Liang et al., 2019).
Поскольку клеточное старение, индуцированное ингибиторами HDAC класса I, сопровождается остановкой деления клеток, но не прекращением их роста и синтеза белков, мы проверили уровень активности mTOR-сигнального пути в клетках mERas, обработанных тубацином, анализируя фосфорилирование мишеней mTOR: содержание фосфорилированного рибосомного белка S6 (p-S6) и фосфорилированного эукариотического фактора инициации транскрипции 4E-BP1 (p-4E-BP1). Представленные на рис. 5а данные показывают, что тубацин увеличивает фосфорилирование 4E-BP1 через 24 ч, затем уровень p-4E-BP1 сохраняется в течение 72 ч, но содержание его изоформ меняется. Что касается S6, то тубацин слабо способствует его фосфорилированию через 24 ч, которое резко снижается к 72 ч (рис. 5а). Таким образом, длительное воздействие тубацина снижает активность mTOR и в то же время вызывает клеточное старение, что отличается от результатов, полученных с бутиратом натрия (Romanov et al., 2010), который и индуцирует старение, и активирует mTOR-путь.
Рис. 5.
Тубацин (Туб) индуциует фосфорилирование белков AMPKα, ULK1 (зависимое от AMPK) и 4E-BP1 – мишени mTOR. а – Иммуноблот с антителами к фосфорилированным формам белков 4E-BP1 (p-4E-BP1) и S6 (p-S6), а также к нефосфорилированному белку S6 после электрофореза клеточных экстрактов (50 мкг) в 10%-ном ПААГ. Клетки обрабатывали или не обрабатывали (−, контроль) 5 мкМ Туб в течение 24 или 72 ч; позиции маркерных белков указаны справа; отн. значения денситометрии полос p-S6/S6 показаны между панелями. б – Иммуноблот с использованием антител к α-тубулину и белку ULK1, фосфорилированному по серину757 (S757) или 555 (S555), после электрофореза экстрактов клеток mERas в 10%-ном ПААГ. Клетки обрабатывали или не обрабатывали (−, контроль) 10 мкM Туб в течение 24 или 72 ч; указано положение маркерного белка 148 кД; отн. значения денситометрии полос р-ULK1/α-тубулин показаны между панелями. в – Иммуноблот с антителами к AMPKα, фосфорилированному по треонину-172 (р-AMPK), и к α-тубулину после электрофореза экстрактов клеток mERas в 10% ПААГ; клетки обрабатывали или не обрабатывали (−) 10 мкM Туб в течение 24, 48 и 72 ч; отн. значения денситометрии полос р-АМPК/α-тубулин показаны между панелями.

Поскольку мы показали модуляцию уровней mTOR в клетках mERas, обработанных тубацином, важно выяснить, как изменяется уровень аутофагии в клетках. Хорошо известно, что высокая активность mTOR негативно регулирует аутофагию посредством ингибиторного фосфорилирования белка ULK-1 (Atg-1) с сопутствующими изменениями двух маркерных белков аутофагии: LC3-II, связанного с мембранами аутофагосом (Kuma et al., 2007), и накоплением p62/SQSTM1, белка секвестосомы 1, (Bjørkøy et al., 2006; Sahani et al., 2014), который может взаимодействовать с HDAC6 и регулировать ее деацетилазную активность (Yan et al., 2013).
Действительно, белок р62 значительно накапливается при обработке тубацином (рис. 6в). Подобное накопление белка р62 в цитоплазме было подтверждено с помощью иммунофлуоресценции с исползованием антител к р62 (данные не показаны). Здесь мы показали, что тубацин индуцировал старение клеток mERas (рис. 4а), а ранее были получены данные о том, что в стареющих фибробластах могут накапливаться агрегаты белка p62 (Gamerdinger et al., 2009). Хотя обычно уровень p62 обратно коррелирует с уровнем аутофагии, но это относится к полному циклу аутофагии. Так, в нормальных MEF и трансформированных клетках HepG2 белок p62 накапливается во время развития аутофагии (Sahani et al., 2014). В клеточных линиях PC3 и LNCaP рака простаты тубацин вызывал накопление p62 благодаря непосредственной регуляции его стабильности независимо от стадии аутофагии (Watson et al., 2016). Поскольку белок р62 физически связан с HDAC6 в цитоплазме, он может модулировать ее деацетилазную активность, так что накопление р62 возможно снижает активность HDAC6 (Yan et al., 2013). В целом, содержание p62 модулируется различными стимулами: окислительным стрессом, активностью киназных каскадов Ras/MAPK и JNK, индуктором аутофагии ресвератролом (Liu et al., 2016). Например, ресвератрол активирует экспрессию p62 через JNK-путь и инициирует аутофагию в клетках K562 (Puissant et al., 2012).
Рис. 6.
Тубацин (Туб) увеличивает количество аутофагосомных вакуолей (АВ) в клетках mERas. а – Количество АВ в контроле (К) и при действии рапамицина (индуктора аутофагии; Рап, 0.5 мкМ) и Туб (5 мкМ) в течение 24 ч; значения относительно контроля (К), принятого за 1 ед., показаны внутри столбцов; контрольные клетки культивировали с растворителем реагентов ДМСО; суспендированные клетки (5–7 × 105) добавляли к компонентам флуоресцентного набора для обнаружения аутофагии CYTO-ID® и анализировали в проточном цитометре (см. раздел “Материал методика”). б – Иммуноблот с антителами к белкам LC3 (формы I и II) и α-тубулину после электрофореза 50 мкг экстрактов клеток в 15%-ном П-ААГ; показаны клетки контрольные (−) и обработанные 5 мкМ Туб 24, 48 и 72 ч; относительные значения денситометрии полос LC3-II/α-тубулина указаны между панелями. в – Иммуноблот с антителами к убиквитин-связывающему белку SQSTM1/p62 (р62) и белку S6 после электрофореза экстрактов клеток в 10%-ном ПААГ; клетки обрабатывали 5 мкМ Туб 24 и 72 ч; (−) – контрольные клетки; значения относительной денситометрии полос p62/S6 указанымежду панелями.

Мы также проверили уровень фосфорилирования белка ULK1 – мишени для киназы AMPK. Тубацин вызывает, главным образом, AMPK-зависимое фосфорилирование по серину-555 (S555), тогда как фосфорилирование по S757 (мишени mTOR) лишь слегка повышено (рис. 5б). Это означает, что AMPK-зависимое фосфорилирование ULK1 (по S555) способствует активации аутофагии (Egan et al., 2011), тогда как mTOR-зависимое фосфорилирование по S757 слабо ингибирует аутофагию (Kim et al., 2011). Кроме того, тубацин способен непосредственно активировать киназу AMPKα (Hawley et al., 1996), усиливая ее фосфорилирование по Thr172 с максимумом через 48 ч (рис. 5в).
Так как ацетилирование α-тубулина необходимо для слияния аутофагосом с лизосомами, HDAC6, вероятно, участвует в контроле процесса аутофагии (Lee et al., 2010). Обработка клеток тубацином в течение 24 ч несильно увеличивает превращение цитоплазматического белка LC3-I в форму LC3-II, связанную с мембранами аутофагосом (рис. 6б), в то время как это превращение хорошо заметно через 72 ч, что свидетельствует об увеличении уровня аутофагии в обработанных тубацином клетках через 3 сут.
Эти результаты согласуются с данными, полученными с помощью набора для аутофагии CytoID (рис. 6а). Можно видеть, что в клетках mERas индуктор аутофагии рапамицин действительно увеличивает количество обнаруженных аутофагосомных вакуолей в среднем в 1.4 раза, в то время как после суточного воздействия тубацина это значение достигает 1.6 раза (рис. 6а). Таким образом, все эти результаты показывают, что тубацин действительно усиливает по крайней мере начальную стадию аутофагии. Подобное действие тубацина на уровень аутофагии также наблюдали в подоцитах (Liang et al., 2020). Эта его роль в сочетании с другими противопухолевыми препаратами может быть перспективной для противораковой терапии (Janku et al., 2011). Так, недавно показали, что комбинация тубацина с эрастином – блокатором транспорта цистеина – индуцирует гибель немезенхимных клеток TNBC рака молочной железы (Alothaim et al., 2021).
Тубацин в сочетании с бортезомибом индуцирует апоптоз. Контрольные клетки mERas показывают высокий уровень протеосомной активности, в частности химотрипсин-подобной активности, которая дополнительно возрастает при действии тубацина (рис. 7а). Поскольку химотрипсин-подобная активность очень чувствительна к бортезомибу, ингибитору протеосомной активности, мы проверили, может ли тубацин в сочетании с бортезомибом вызывать синергический антипролиферативный эффект в клетках mERas. Действительно, данные проточной цитометрии (рис. 7б1) показывают, что тубацин и бортезомиб вместе уменьшают S-фазу клеточного цикла до 18%. Интересно, что отдельно бортезомиб способен индукцировать G1/S-блок в клеточном цикле mERas, причем доля G1-фазы возрастает при увеличении концентраци бортезомиба (рис. 7в1). Индуцированный бортезомибом G1/S-блок также наблюдали в некоторых линиях опухолевых клеток (хондросаркомы, В-лимфомы Раджи) (Bao et al., 2017; Gu et al., 2017), тогда как G2/M-блок цикла был обнаружен в других опухолевых клетках (саркомы Ивинга, рефрактерной B-лимфомы мантии, колоректального рака) (Lu et al., 2008; Hutter et al., 2012; Rastogi, Mishra, 2012).
Рис. 7.
Протеосомный ингибитор бортезомиб (Бз) усиливает индуцированный тубацином G1/S-блок клеточного цикла и в сочетании с тубацином (Туб, 5 мкМ) способствует апоптотической гибели клеток mERas. а – активность химотрипсин-подобной пептидазы с флуорогенным субстратом (50 мкМ, Enzo) в экстрактах клеток (5 мкг белка) после обработки ингибитором HDAC в течение 72 ч; К – контрольные клетки, Бут – 4 мМ бутират натрия; значения активности пептидазы относительно контроля (К), принятого за 1 ед., показаны внутри столбцов. б1 – Распределение клеток по фазам клеточного цикла после суточной обработки 15 мкМ Туб и (или) 10 нМ Бз по данным проточной цитометрии, (−) – контрольные клетки; указаны средние значения и их ошибки по 3 экспериментам. б2 – доля (в %) апоптотирующих клеток, измеренная по площади пиков субдиплоидной ДНК для одного из экспериментов, представленного на диаграмме б1. в1 – Распределение клеток по фазам клеточного цикла после обработки (24 ч) Бз в концентрации 10, 15 и 20 нМ; (−) – контрольные клетки; указаны средние значения и их ошибки по 3 экспериментам. в2 – Доля (в %) апоптотирующих клеток, измеренная по площади пиков субдиплоидной ДНК для одного из экспериментов, представленного на диаграмме в1.

Супрессорное действие бортезомиба на клеточный цикл можно объяснить индукцией экспрессии ингибиторов клеточного цикла (р21, р27, р15) (Lu et al., 2008; Hutter et al., 2012; Rastogi, Mishra, 2012), а также его способностью препятствовать деградации этих регуляторных белков (Lu et al., 2008; Gu et al., 2017).
При низкой концентрации (10 нМ) бортезомиб не вызывает заметной апоптотической гибели клеток mERas (рис. 7б2, в2). После совместной обработки тубацином и бортезомибом наблюдали дополнительный (синергетический) эффект по снижению доли клеток в S-фазе (рис. 7б1). В то же время, по данным проточной цитометрии в клетках, обработанных тубацином и бортезомибом, наблюдали увеличение субдиплоидного пика как характеристики продолжающейся апоптотической гибели клеток (рис. 7б2). Этот факт можно объяснить значительным снижением жизнеспособности клеток mERas при совместном действии тубацина и бортезомиба, которое выявлялось с помощью набора Count and Viability (рис. 8а). Повышенная апоптотическая гибель клеток mERas в присутствии тубацина и бортезомиба подтверждается проточной цитометрией с использованием набора Аннексин-V-FITC и последующим анализом двупараметрического распределения клеток (рис. 8б, в).
Рис. 8.
Совместное действие тубацина (Туб, 15 мкМ) и бортезомиба (Бз, 15 нМ) значительно снижает жизнеспособность клеток mERas вследствие активации апоптоза. а – Жизнеспособность клеток, оцененная с помощью набора Count and Viability для цитометра Muse Cell Analyzer, (−) – контроль; процентное количество живых клеток указано внутри столбцов. б – Двупараметрическое распределение клеток (содержание ДНК против Аннексин V-FITC) по данным проточной цитометрии; показаны контрольные клетки (К), действие Туб, Бз, раздельно и совместно. в – Диаграмма по результатам одного из экспериментов, представленного в части б этого рисунка; показано процентное содержание апоптотических клеток.

Ранее аналогичный комбинированный эффект был обнаружен для клеток множественной миеломы человека (Hideshima et al., 2005). При использовании другой модели было показано, что комбинация бортезомиба и индуцирующего аутофагию рапамицина может подавлять гепатоцеллюлярную карциному мыши (Wang et al., 2012). В совокупности эти результаты показывают, что ингибитор HDAC6 тубацин усиливает протеосомную активность в клетках mERas, а совместное воздействие тубацина и бортезомиба оказывает дополнительное антипролиферативное действие и вызывает апоптотическую гибель клеток.
Суммируя, можно сказать, что ингибитор HDAC6 тубацин действует через ацетилирование α-тубулина, который является основным компонентом микротрубочек, участвующих в процессе аутофагии. Подобно ингибитору HDAC бутирату натрия, тубацин вызывает остановку клеточного цикла на стадии G1/S, подавляет пролиферацию клеток mERas, а также вызывает их гипертрофию и клеточное старение, что приводит к подавлению миграции клеток в рану. Следует отметить, что тубацин осуществляет все эти эффекты при гораздо более низких концентрациях (микромолярных) по сравнению с бутиратом натрия (4 мМ).
Полученные результаты показывают, что тубацин способен индуцировать по меньшей мере первые стадии аутофагии путем активации как AMPK-зависимого фосфорилирования ULK1, так и самого фосфорилирования AMPKα. Хотя белок LC3-II накапливается в течение 72 ч после обработки клеток тубацином, слияние аутофагосом и лизосом, по-видимому, не происходит. Комбинация протеосомного ингибитора бортезомиба в наномолярных концентрациях и тубацина очень эффективна для индукции апоптотической гибели клеток mERas, что предполагает перспективное использование ее для элиминации Ras-экспрессирующих опухолевых клеток.
Список литературы
Поспелова Т.В., Кислякова Т.В., Медведев А.В., Светликова С.Б., Поспелов В.А. 1990. Характеристика трансформированного фенотипа и экспрессии САТ-плазмид в клетках эмбриональных фибробластов крысы, трансформированных онкогенами E1A + cHa-ras. Цитология. Т. 32. С. 148. (Pospelova T.V., Kislyakova T.V., Medvedev A.V., Svetlikova S.B., Pospelov V.A. 1990. The characteristics of the transformed phenotype and the expression of indicator plasmids in the cells of rat embryonic fibroblasts immortalized by oncogene E1Aad5 and transformed by oncogenes E1Aad5+c-Ha-ras. Tsitologiya. V. 32. P. 148.)
Abramova M.V., Pospelova T.V., Nulenkov F.P., Hollander C.V., Fornace A.J. Jr., Pospelov V.A. 2006. G1/S arrest induced by histone deacetylase inhibitor sodium butyrate in E1A + Ras-transformed cells is mediated through downregulation of E2F activity and stabilization of beta-catenin. J. Biol. Chem. V. 281. P. 21040.
Adams J.M., Cory S. 2007. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene.V. 26. P. 1324.
Aldana-Masangkay G.I., Rodrigues-Gonzales A., Lin T., Ikeda A.K. Hsieh Y.-T., Kim Y.-M., Lomenick B., Okemoto K., Landaw E.M., Wang D., Mazitschek R., Bradner J.E., Sakamoto K.M. 2011. Tubacin suppresses proliferation and induces apoptosis of acute lymphoblastic leukemia cells. Leuk. Lymphoma. V. 52. P. 1544.
Alothaim T., Charbonneau M., Tang X. 2021. HDAC6 inhibitors sensitize non-mesenchymal triple-negative breast cancer cells to cysteine deprivation. Sci. Rep. V. 11. P. 10956.
Asthana J., Kapoor S., Mohan R., Panda D. 2013. Inhibition of HDAC6 deacetylase activity increases its binding with microtubules and suppresses microtubule dynamic instability in MCF-7 cells. J. Biol. Chem. V.288. P. 22516.
Bao X., Ren T., Huang Y., Ren C., Yang K., Zhang H., Guo W. 2017. Bortezomib induces apoptosis and suppresses cell growth and metastasis by inactivation of Stat3 signaling in chondrosarcoma. Int. J. Oncol. V. 50. P. 477.
Bjørkøy G., Lamark T., Johansen T. 2006. p62/SQSTM1: a missing link between protein aggregates and the autophagy machinery. Autophagy. V. 2. P. 138.
Bradford M.M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analyt. Biochem. V. 72. P. 248.
Davie J.R. 2003. Inhibition of histone deacetylase activity by butyrate. J. Nutr. V. 133. P. 2485.
Delbridge A.R., Strasser A. 2015. The BCL-2 protein family, BH3-mimetics and cancer therapy. Cell Death Differ. V. 22. P. 1071.
Depetter Y., Geurs S., De Vreese R., Goethals S. Vandoom E., Steenbrugge J., Meyer E., de Tullio P., Brake M., D’hooghe M., de Weyer O. 2019. Selective pharmacological inhibitors of HDAC6 reveal biochemical activity but functional tolerance in cancer models. Int. J. Cancer. V. 145. P. 735.
Deskin B., Yin Q., Saito S., Shan B., Lasky J.A. 2020. Inhibition of HDAC6 attenuates tumor growth of non-small cell lung cancer. Transl Oncol. V. 13. P. 135.
Dimri G.P., Lee X., Basile G., Acosta M., Scott G., Roskelley C., Medrano E.E., Linskens M., Rubel, I., Pereira-Smith O. 1995. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA. V. 92. P. 9363.
Ding N., Ping L., Feng L., Zheng X., Song Y., Zhu J. 2014. Histone deacetylase 6 activity is critical for the metastasis of Burkitt’s lymphoma cells. Cancer Cell Int. V. 14. P. 139.
Egan D.F., Shackelford D.B., Mihaylova M.M., Gelino S., Kohnz R.A., Mair W., Vasquez D.S., Joshi A., Gwinn D.M., Taylor R., Asara J.M., Fitzpatrick J., Dillin A., Violle, B., Kundu M., Hansen M., Shaw R.J. 2011. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. V. 331. P. 456.
Gamerdinger M., Hajieva P., Kaya A.M., Wolfrum U., Hart F.U., Behl C. 2009. Protein quality control during aging involves recruitment of the macroautophagy pathway by BAG3. EMBO J. V. 28. P. 889.
Gu J.J., Kaufman G.P., Mavis C., Czuczman M.S., Hernandez-Ilizaliturri F.J. 2017. Mitotic catastrophe and cell cycle arrest are alternative cell death pathways executed by bortezomib in rituximab resistant B-cell lymphoma cells. Oncotarget. V. 8. P. 12741.
Guardiola A.R., Yao T.-P. 2002. Molecular cloning and characterization of a novel histone deacetylase HDAC10. J. Biol. Chem. V. 277. P. 3350.
Haggarty S.J., Koeller K.M., Wong J.C., Grozinger C.M., Schreiber S.L. 2003. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc. Natl. Acad. Sci. USA. V. 100. P. 4389.
Hawley S.A., Davison M., Woodsi A., Davies S.P., Beri R.K., Carling D., Hardie D.G. 1996. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J. Biol. Chem. V. 271. P. 27879.
Hideshima T., Bradner J.E., Wong J., Chauhan D., Richardson P., Schreiber S.L., Anderson K.C. 2005. Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proc. Natl. Acad. Sci. USA. V. 102. P. 8567.
Hutter G., Rieken M., Pastore A. 2012. The proteasome inhibitor bortezomib targets cell cycle and apoptosis, and acts synergistically in a sequence-dependent way with chemotherapeutic agents in mantle cell lymphoma. Ann. Hematol. V. 91. P. 847.
Janku F., McConkey D.J., Hong D.S., Kurzrock R. 2011. Autophagy as a target for anticancer therapy. Nat. Rev. Clin. Oncol. V. 8. P. 528.
Kim J., Kundu M., Violle, B., Guan K.L. 2011. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. V. 13. P. 132.
Kulichkova V.A., Tsimokha A.S., Fedorova O.A., Moiseeva T.N., Bottril A., Lezina L., Gauze L.N., Konstantinova I.M., Mitytenberg A.G., Barlev N.A. 2010. 26S proteasome exhibits endoribonuclease activity controlled by extra-cellular stimuli. Cell Cycle. V. 9. P. 840.
Kuma A., Matsui M., Mizushima N. 2007. LC3, an autophagosome marker, can be incorporated into protein aggregates independent of autophagy. Autophagy. V. 3. P. 323.
Laberge R.M., Sun Y., Orjalo A.V., Patil C.K., Freund A., Zhou L., Curran S.C., Davalos A.R., Wilson-Edell K.A., Liu S., Limbad C., Demaria M., Li P., Hubbard G.B., Ikeno Y. et al. 2015. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat. Cell Biol. V. 17. P. 1049.
Lee J.Y., Koga H., Kawaguchi Y., Tang W., Wong E., Gao Y.-S., Pandey U.B., Kayshik S., Tresse E., Lu J., Taylor J.P., Cuervo A.M., Yao T.-P. 2010. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. V. 29. P. 969.
Liang J.-Q., Lu F., Gan B., Wen Y.-Y., Chen J., Wang H.-G., Yang Y., Peng X.-S., Zhou Y.-F. 2019. Low-dose tubacin promotes BMSCs proliferation and morphological changes through the ERK pathway. Am. J. Transl. Res. V. 11. P. 1446.
Liang T., Qi C., Lay Y., Xie J., Wang H., Zhang L., Lin T., Jv M., Li J., Wang Y., Zhang Y., Chen Z., Qiu X., Li R., Li Z. et al. 2020. HDAC6-mediated α-tubulin deacetylation suppresses autophagy and enhances motility of podocytes in diabetic nephropathy. J. Cell Mol. Med. V. 24. P. 11558.
Liu W.J., Ye L., Huang W.F., Guo L.J., Xu Z.G., Wu H.L., Yang C., Liu H.F. 2016. P62 links the autophagy pathway and the ubiqutin–proteasome system upon ubiquitinated protein degradation. Cell. Mol. Biol. Lett. V. 21. P. 1.
Lu G., Vasu P.V., Chaudhary P.M. 2008. Proteasome inhibitor bortezomib induces cell cycle arrest and apoptosis in cell lines derived from Ewing’s sarcoma family of tumors and synergizes with TRAIL. Cancer Biol. Therapy. V. 7. P. 603.
Mathew R., Karantza-Wadsworth V., White E. 2007. Role of autophagy in cancer. Nat. Rev. Cancer. V. 7. P. 961.
Matsuyama A., Shimazu T., Sumida Y., Saito A., Yoshimatsu Y., Seigneurin-Berny D., Osada H., Komatsu Y., Nishino N., Khochbin S., Horinouchi S., Yoshida M. 2002. In vivo destabilization of dynamic microtubules by HDAC6-mediated deacetylation. EMBO J. V. 21. P. 6820.
Namdar M., Perez G., Ngo L., Marks P.A. 2010. Selective inhibition of histone deacetylase 6 (HDAC6) induces DNA damage and sensitizes transformed cells to anticancer agents. Proc. Natl. Acad. Sci. USA. V. 107. P. 20003.
Nelyudova A.M., Aksenov N.D., Pospelov V.A., Pospelova T.V. 2007. By blocking apoptosis, Bcl-2 in p38-dependent manner promotes cell cycle arrest and accelerated senescence after DNA damage and serum withdrawal. Cell Cycle. V. 6. P. 2171.
Newbold A., Falkenberg K.J., Prince H.M., Johnstone R.W. 2016. How do tumor cells respond to HDAC inhibition? FEBS J. V. 283. P. 4032.
Puissan, A., Fenouille N., Auberger P. 2012. When autophagy meets cancer through p62/SQSTM1. Am. J. Cancer Res. V. 2. P. 397.
Rastogi N., Mishra D.P. 2012. Therapeutic targeting of cancer cell cycle using proteasome inhibitors. Cell Division. V. 7. P. 1.
Rodier F., Campisi J. 2011. Four faces of cellular senescence. J. Cell Biol. V. 192. P. 547.
Romanov V.S., Abramova M.V., Svetlikova S.B., Bykova T.V., Zubova S.G., Aksenov N.D., Fornace A.J.Jr., Pospelova T.V., Pospelov V.A. 2010. p21Waf1 is required for cellular senescence but not for cell cycle arrest induced by the HDAC inhibitor sodium butyrate. Cell Cycle. V. 9. P. 3945.
Sahani M.H., Itakura E., Mizushima N. 2014. Expression of the autophagy substrate SQSTM1/p62 is restored during prolonged starvation depending on transcriptional upregulation and autophagy-derived amino acids. Autophagy. V. 10. P. 431.
Shao Y., Gao Z., Marks P.A., Jiang X. 2004. Apoptotic and autophagic cell death induced by histone deacetylase inhibitors. Proc. Natl. Acad. Sci. USA. V. 101. P. 18030.
Sobue S., Mizutani N., Aoyama Y., Kawamoto Y., Suzuki M., Nozawa Y., Ichihara M., Murate T. 2016. Mechanism of paclitaxel resistance in a human prostate cancer cell line, PC3-PR, and its sensitization by cabazitaxel. Biochem. Biophys. Res. Comm. V. 479. P. 808.
Verdel A., Curtet S., Brocard M., Rousseaux S., Lemercier C., Yoshida M., Khochbin S. 2000. Active maintenance of mHDA2/mHDAC6 histone-deacetylase in the cytoplasm. Curr. Biol. V. 10. P. 747.
Wang C., Gao D., Guo K., Kang X., Jiang K., Sun C., Li Y., Sun L., Shu H., Jin G., Sun H., Wu W., Liu Y. 2012. Novel synergistic antitumor effects of rapamycin with bortezomib on hepatocellular carcinoma cells and orthotopic tumor model. BMC Cancer. V. 12. P. 1.
Watson G.W., Wickramasekara S., Fang Y., Maier C.S., Williams D.E., Dashwood R.H., Perez V.I., Ho E. 2016. HDAC6 activity is not required for basal autophagic flux in metastatic prostate cancer cells. Exp. Biol. Med. V. 241. P. 1177.
Yan J., Seibenhener M.L., Calderilla-Barbosa L., Diaz-Meco M.-T., Moscat J., Jiang J., Wooten M.W., Wooten M.C. 2013. SQSTM1/p62 interacts with HDAC6 and regulates deacetylase activity. PLoS one. V. 8. P. 1.
Yu S., Cai X., Wu C., Liu Y., Zhang J., Gong X., Wang X., Wu X., Zhu T., Mo L., Gu J., Yu Z., Chen J., Thiery J.P., Chai R., Chen L. 2017. Targeting HSP90-HDAC6 regulating network implicates precision treatment of breast cancer. Int. J. Biol. Sci. V. 13. P. 505.
Zhang Y., Li N., Caron C., Matthias G., Hess D., Khochbin S., Matthias P.2003. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. V. 22. P. 1168.
Дополнительные материалы отсутствуют.