

Журнал аналитической химии, 2020, T. 75, № 1, стр. 67-73
Хромато-масс-спектрометрическое определение 1-гидроксипирена в моче как биомаркера воздействия полициклических ароматических углеводородов
А. Н. Алексеенко a, *, О. М. Журба a, А. В. Меринов a, С. Ф. Шаяхметов a
a Восточно-Сибирский институт медико-экологических исследований
665827 Ангарск, Россия
* E-mail: alexeenko85@mail.ru
Поступила в редакцию 01.10.2018
После доработки 10.11.2018
Принята к публикации 29.03.2019
Аннотация
Разработана простая и чувствительная методика определения 1-гидроксипирена в моче как биологического маркера воздействия полициклических ароматических углеводородов. После ферментативного гидролиза для расщепления коньюгата аналит извлекали из матрицы жидкостно–жидкостной экстракцией н-гексаном, экстракт упаривали до сухого остатка и дериватизировали аналит силилирующим реагентом N,O-бис(триметилсилил)трифторацетамидом в триметилсилиловый эфир при комнатной температуре. Триметилсилильный экстракт анализировали капиллярной газовой хроматографией с масс-селективным детектированием. Количественное высокоточное определение достигнуто за счет использования изотопно-меченного стандарта 1-гидроксипирена-d9. Матричных эффектов не наблюдали. Предел обнаружения 0.02 нг/мл, линейный диапазон 0.1–100 нг/мл. Относительные стандартные отклонения в условиях повторяемости и внутрилабораторной прецизионности 0.044 и 0.064 соответственно. Процентная мера правильности 96–102%.
Полициклические ароматические углеводороды (ПАУ), обладая высокой канцерогенной и мутагенной активностью, представляют серьезную угрозу здоровью человека и его потомству. В связи с высокой опасностью ПАУ в России и в большинстве других стран проводится мониторинг наиболее опасного бенз(а)пирена в объектах окружающей среды и в пищевых продуктах. Альтернативный способ оценки влияния ПАУ на человека основан на определении их метаболитов в биологических средах, однако его применяют только в развитых странах.
Концентрация остаточных, не превратившихся в метаболиты количеств ПАУ как в кровеносной системе, так и в моче человека настолько мала, что с трудом поддается измерению современными аналитическими методами. Зато уровень содержания некоторых метаболитов достаточно высок. Самой обычной и легкодоступной биологической средой при определении метаболитов ПАУ в организме человека является моча. В подавляющем случае ПАУ присутствуют в виде смесей, причем одним из основных компонентов по уровню концентрации является пирен. Метаболит пирена 1-гидроксипирен – стабильное соединение, относительно легко и надежно определяемое в моче. На основании большого количества исследований можно с достаточной уверенностью признать 1-гидроксипирен адекватным биомаркером присутствия ПАУ в окружающей среде [1–3].
Для определения 1-гидроксипирена в моче применяют ВЭЖХ с флуориметрическим детектированием [4]. Пробоподготовка включает предварительный ферментативный гидролиз β-глюкуронидазой при 37°С в течение 16 ч, твердофазную экстракцию (ТФЭ) на патроне С18, упаривание элюата досуха и растворение остатка в метаноле. Предел обнаружения 0.1 нг/мл, нижняя граница определяемых содержаний 2 нг/мл при объеме пробы 10 мл, относительное стандартное отклонение sr (воспроизводимость) 0.126, коэффициент корреляции градуировочного графика 0.990, процентная мера правильности 88 ± 9%.
Другой подходящий метод определения 1-гидроксипирена в моче – газовая хроматография с масс-селективным детектированием (ГХ–МС) в режиме электронной ионизации (ЭИ) [5, 6]. Основное преимущество метода ГХ–МС по сравнению с ВЭЖХ – это лучшее разделение компонентов и возможность применения изотопно-меченного стандарта d9-1-гидроксипирен (дейтерированный стандарт). Для метода ГХ–МС 1-гидроксипирен, как и его дейтерированный аналог, необходимо дериватизировать, так как наличие OH-группы обусловливает низкую летучесть, низкую чувствительность и может вызвать взаимодействие аналита с неподвижной фазой колонки. Наиболее распространенными реакциями дериватизации для веществ, содержащих гидроксильные группы, являются ацилирование и силилирование [7, 8]. В реакциях ацилирования соединения, содержащие подвижные атомы водорода, превращают в сложные эфиры при действии карбоновой кислоты или ее ангидридов. Из-за присутствия остаточных количеств кислоты продукты нельзя непосредственно вводить в газовый хроматограф, и требуется этап очистки. В реакциях силилирования подвижные атомы водорода заменяют триметилсилильной или трет-бутилдиметилсилильной группой [9]. Продукты этой реакции, как правило, более летучие и термически стабильные. В отличие от ацилирования, силилирование обычно не требует стадии очистки, и производные могут быть введены непосредственно в ГХ. Силилирование – наиболее распространенный метод дериватизации [9], а классическими реагентами для этого являются триметилхлорсилан (ТМХС), триметилэтилсилилимидазол, N-метилтриметилсилилтрифтороацетамид, N,O-бис(триметилсилил)трифторацетамид (БСТФА) и N-(трет-бутилдиметилсилил)-N-метилтрифторацетамид, из которых наиболее часто используют два последних [10].
Определение методом ГХ–МС включает следующие стадии [5, 6, 10]: ферментативный гидролиз β-глюкуронидазой при 37°С в течение 17–18 ч, ТФЭ, упаривание экстракта в токе азота, дериватизацию сухого остатка силилирующим реагентом БСТФА при 60°С в течение 40 мин, анализ методом ГХ–МС. Продолжительность разделения 40 мин. Предел обнаружения 0.5 нг/мл, предел определения 1 нг/мл при объеме пробы 3 мл, sr составляет 0.067–0.131, коэффициент корреляции градуировочного графика 0.995, процентная мера правильности 88.7%.
Цель настоящей работы – разработка методики определения 1-гидроксипирена в моче методом ГХ–МС с использованием изотопно-меченного стандарта. Для этого решали следующие задачи: повышение чувствительности определения за счет увеличения степени экстракции из биологической матрицы; значительное сокращение времени пробоподготовки за счет уменьшения продолжительности ферментативного гидролиза и дериватизации 1-гидроксипирена силилирующим реагентом БСТФА.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Оборудование. Газовый хроматограф Agilent 7890A с масс-селективным детектором Agilent 5975С (ЭИ), автоматическим дозатором Agilent 7693, капиллярной колонкой HP-5MS (30 м × × 0.25 мм, 0.25 мкм). Блочный термостат Stuart (30–130°С), вибросмеситель Biosan, центрифуга Eppendorf 5804, система упаривания в токе азота.
Реактивы и стандарты. 1-Гидроксипирен (99.8%, Aldrich); 1-гидроксипирен-d9 (98.8%, Santa Cruz); ацетонитрил (Криохром); водный раствор β-глюкуронидазы Helix Pomatia H-2 (>85 000 ед./мл, Aldrich); уксусно-ацетатный буферный раствор с рН 5; сульфат магния х. ч.; н-гексан (Криохром); БСТФА, содержащий 1% ТХМС (Fluka).
Пробоподготовка. К 2 мл мочи в центрифужной пробирке емк. 15 мл добавляли 20 мкл раствора 1-гидроксипирена-d9 в ацетонитриле (5 мкг/мл), 1 мл ацетатно-уксусного буферного раствора с рН 5, 20 мкл водного раствора β-глюкуронидазы и нагревали 1 ч при 55°С, затем раствор охлаждали в течение 20 мин до комнатной температуры, добавляли 0.5 г сульфата магния, 2 мл н-гексана и интенсивно встряхивали на вибросмесителе в течение 2 мин, разделяли фазы центрифугированием при 4000 об/мин в течение 10 мин, отделяли гексановый экстракт, перенося его во флакон емк. 5 мл с коническим дном, снова добавляли 2 мл н-гексана, повторяли процесс, экстракты объединяли. Экстракт упаривали досуха в небольшом токе азота при температуре водяной бани 60°С, затем к сухому остатку добавляли 100 мкл реагента БСТФА и выдерживали 5 мин, после чего переносили в стеклянную вставку в микрофлакон емк. 200 мкл и анализировали.
Хромато-масс-спектрометрия. Определение осуществляли в следующих условиях: температура испарителя 300°С; объем водимой пробы 1 мкл; режим ввода образца без деления потока 0.7 мин; линейная скорость в колонке 40 см/с; режим термостата колонки: выдержка при 60°С в течение 2 мин, подъем со скоростью 15 град/мин до 300°С, выдержка в течение 2 мин; температура интерфейса 290°С, ионного источника ЭИ 230°С, квадруполя 150°С; задержка включения нити накала 16.6 мин. Сбор хромато-масс-спектрометричекой информации осуществляли в режиме мониторинга выделенных ионов с m/z 290, 275, 299, 284.
1-Гидроксипирен и 1-гидроксипирен-d9 в виде производных (триметилсиланов) идентифицировали на хроматограммах по времени удерживания и соотношению интенсивностей регистрируемых ионов (табл. 1).
Таблица 1.
Данные для идентификации на масс-хроматограммах 1-гидроксипирена и его изотопно-меченного стандарта
| Соединение | Время удерживания, мин | m/z (основной) | m/z (подтверждающий) |
|---|---|---|---|
| 1-Гидроксипирен-d9 (внутренний стандарт) | 17.14 | 299 (100%) | 284 (20 ± 5%) |
| 1-Гидроксипирен | 17.16 | 290 (100%) | 275 (20 ± 5%) |
Для количественного определения использовали метод внутреннего стандарта по растворам с концентрацией 1-гидроксипирена в моче с 0.1 до 100 нг/мл.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Силилирование N,O-бис(триметилсилил)трифторацетамидом. Сравнили силилирование 1-гидроксипирена реагентом БСТФА при комнатной температуре (20–25°С) и при 90°С и различной продолжительности реакции (табл. 2). Видно, что при силилировании температура и продолжительность реакции не оказывают значимого влияния на аналитический сигнал, особенно если в качестве последнего выступает относительная площадь (высота) пика. Силилирование достаточно проводить при комнатной температуре в течение 5 мин с изотопно-меченным стандартом, при этом значение sr минимально.
Таблица 2.
Зависимость степени дериватизации 1-гидроксипирена реагентом N,O-бис(триметилсилил)трифторацетамидом от времени реакции и температуры
| Время реакции, мин | Sабс* | Sотн** | ||
|---|---|---|---|---|
| 20–25оС | 90оС | 20–25оС | 90оС | |
| 5 | 3150 | 3492 | 0.38 | 0.34 |
| 30 | 3039 | 3065 | 0.39 | 0.36 |
| 45 | 3419 | 3174 | 0.37 | 0.33 |
| sr | 0.061 | 0.068 | 0.026 | 0.044 |
Выбор способа извлечения 1-гидроксипирена из пробы мочи. Установлено, что при ТФЭ степень извлечения 1-гидроксипирена из мочи составляет 8%, а при жидкостно–жидкостной экстракции (ЖЖЭ) н-гексаном − около 90% (табл. 3).
Таблица 3.
Извлечение 1-гидроксипирена из водной и биологической матриц разными способами
| Способ извлечения | Матрица | Условия | R, % |
|---|---|---|---|
| ТФЭ | Моча | Сорбент С18, элюент – метанол | 8 |
| Вода | 88 | ||
| ЖЖЭ | Моча | Двукратная экстракция гексаном | 90 |
Несмотря на то, что ТФЭ − более современный способ экстрагирования по сравнению с ЖЖЭ, степень извлечения аналита ТФЭ очень низкая. Это можно объяснить главным образом тем, что в образце мочи присутствует большое количество полярных соединений, содержащих в своем составе OH–, COOH–, NH=, −NH2-группы, причем в бóльших концентрациях, чем аналит. Эти компоненты оказывают мешающее влияние при силилировании 1-гидроксипирена реагентом БСТФА, поэтому в реакцию силилирования вступает очень малое количество 1-гидроксипирена. При ЖЖЭ гексаном извлекаются соединения неполярной или малополярной природы, которые не мешают дериватизации. Оптимальным способом извлечения 1-гидроксипирена из биологической матрицы является двукратная ЖЖЭ гексаном.
Выбор условий жидкостно–жидкостной экстракции 1-гидроксипирена с помощью математического планирования. Основными факторами, влияющими на эффективность ЖЖЭ, являются тип экстрагента, продолжительность экстракции, число экстракций, природа и количество высаливающего агента. Установлено, что н-гексан является более эффективным экстрагентом, чем диэтиловый эфир и толуол, благодаря снижению мешающих влияний при дериватизации. В качестве высаливающего агента лучше применять сульфат магния, а не сульфат натрия, так как он обеспечивает более высокую степень извлечения аналита. С помощью математического планирования [11] оптимизировали следующие количественные параметры ЖЖЭ 1-гидроксипирена: массу сульфата магния, продолжительность экстракции, кратность экстракции (табл. 4).
Таблица 4.
Условия планирования трехфакторного эксперимента
| Фактор | Нулевой уровень x0 | Интервал варьирования J | Нижний уровень фактора | Верхний уровень фактора |
|---|---|---|---|---|
| x1 – масса MgSO4, г | 0.5 | 0.5 | 0 | 1 |
| x2 – время встряхивания, мин | 3 | 2 | 1 | 5 |
| x3 – кратность экстракции (качественный фактор) | – | – | 1 | 2 |
В многофакторном эксперименте одновременно варьировали три фактора. Матрица планирования состояла из 8 опытов. За параметр оптимизации y приняли степень экстракции аналита. Каждый опыт в матрице планирования (табл. 5) повторяли трижды.
Таблица 5.
Матрица планирования для проведения трехфакторного эксперимента и полученные значения параметра оптимизации
| № опыта | Факторы | Параметр оптимизации yj (n = 3) |
|||||
|---|---|---|---|---|---|---|---|
| натуральные | кодированные | ||||||
| x1 | x2 | x3 | x1 | x2 | x3 | ||
| 1 | 0 | 1 | 1 | – | – | – | 80.3 |
| 2 | 1 | 1 | 1 | + | – | – | 73.2 |
| 3 | 0 | 5 | 1 | – | + | – | 71.6 |
| 4 | 1 | 5 | 1 | + | + | – | 80.0 |
| 5 | 0 | 1 | 2 | – | – | + | 93.0 |
| 6 | 1 | 1 | 2 | + | – | + | 93.4 |
| 7 | 0 | 5 | 2 | – | + | + | 89.8 |
| 8 | 1 | 5 | 2 | + | + | + | 90.2 |
Статистическим путем получили уравнение:
Из уравнения (1) видно, что кратность экстракции (x3) вносит больший вклад в формирование отклика (степень экстракции), чем масса сульфата магния и время экстракции (встряхивание в вибросмесителе). Степень экстракции аналита выше при двукратной экстракции вследствие увеличения концентрации аналита в органической фазе. Подставляя в уравнение кодированные значения факторов, рассчитали теоретические значения параметра оптимизации, которые показывают, что степень экстракции не меняется (табл. 6). Следовательно, нет смысла увеличивать массу сульфата магния x1 и время экстракции x2. Массу сульфата магния целесообразно зафиксировать на нулевом уровне, а время экстракции уменьшить до 2 мин. Таким образом, оптимальные условия ЖЖЭ 1-гидрокиспирена из мочи следующие: масса сульфата магния 0.5 г, время экстракции 1–2 мин, двукратная экстракция.
Таблица 6.
Условия постановки опытов и теоретические значения параметра оптимизации
| № опыта | Натуральные значения факторов | Кодированные значения факторов | y | ||||
|---|---|---|---|---|---|---|---|
| x1 | x2 | x3 | x1 | x2 | x3 | 92.0 | |
| 1 | 0.5 | 1 | 2 | 0 | –1 | 1 | 92.0 |
| 2 | 0.5 | 2 | 2 | 0 | –0.5 | 1 | 92.0 |
| 3 | 0.5 | 3 | 2 | 0 | 0 | 1 | 92.0 |
| 4 | 0.5 | 4 | 2 | 0 | 0.5 | 1 | 92.0 |
| 5 | 0.5 | 5 | 2 | 0 | 1 | 1 | 92.0 |
| 6 | 0.6 | 3 | 2 | 0.20 | 0 | 1 | 92.0 |
| 7 | 0.8 | 3 | 2 | 0.60 | 0 | 1 | 92.0 |
| 8 | 1 | 3 | 2 | 1.00 | 0 | 1 | 92.0 |
Ферментативный гидролиз. В табл. 7 приведены результаты анализа реальных образцов мочи работников алюминиевого производства с использованием двух вариантов гидролиза β-глюкуронидазой. Видно, что концентрации 1-гидроксипирена, полученные двумя способами, различаются не сильно. Во втором варианте концентрации аналита выше, поэтому ферментативный гидролиз β-глюкуронидазой лучше проводить при 55°С в течение 60 мин.
Таблица 7.
Концентрация (нг/мл) 1-гидроксипирена в моче работников алюминиевого производства при разных вариантах ферментативного гидролиза β-глюкуронидазой
| № образца мочи | Условия ферментативного гидролиза β-глюкоронидазой | |
|---|---|---|
| 37оС, 16 ч | 55оС, 1 ч | |
| 1 | 0.5 | 0.6 |
| 2 | 1.2 | 1.5 |
| 3 | 2.7 | 2.9 |
| 4 | 2.4 | 2.5 |
| 5 | 34.5 | 39 |
| 6 | 0.83 | 0.96 |
| 7 | 0.31 | 0.35 |
| 8 | 22.3 | 25 |
Валидация методики. Оценены предел обнаружения, предел определения, линейный диапазон, повторяемость, внутрилабораторная прецизионность, правильность, матричный эффект, селективность [12].
Предел обнаружения, рассчитанный для соотношения сигнал/шум ≥3, составил 0.02 нг/мл, предел определения (соотношение сигнал/шум ≥10) – 0.1 нг/мл. Линейный диапазон установили по 6 модельным образцам мочи с разными концентрациями 1-гидроксипирена (0.1, 2, 10, 20, 40, 100 нг/мл), коэффициент корреляции r > 0.999. Приемлемое значение r не ниже 0.990. Таким образом, линейный диапазон составил от 0.1 до 100 нг/мл. Точность (прецизионность и правильность) оценивали для 4 образцов мочи с добавками аналита 0.5, 2, 20 и 40 нг/мл, каждый образец анализировали дважды в течение пяти дней. По результатам анализа рассчитывали значения sr (повторяемость и внутрилабораторная прецизионность) и процентную меру правильности как отношение значений найденной и введенной концентрации (табл. 8). Все отношения, лежащие в диапазоне от 96.5 до 102% от номинальной концентрации определяемого вещества, соответствовали приемлемому критерию (100 ± 7%). Значения повторяемости и внутрилабораторной прецизионности удовлетворяют приемлемым критериям (не выше 12%). Введенные концентрации попадают в доверительные интервалы найденных значений.
Таблица 8.
Прецизионность и правильность определения 1-гидроксипирена в моче (P = 0.95)
| Введено, нг/мл | Найдено, нг/мл | Внутрилабораторная прецизионность, sr (n = 5) |
Повторяемость, sr (n = 2) | Правильность, % |
|---|---|---|---|---|
| 0.5 | 0.51 ± 0.04 | 0.063 | 0.044 | 102 |
| 2 | 1.9 ± 0.1 | 0.064 | 0.010 | 97 |
| 20 | 19 ± 1 | 0.064 | 0.017 | 96.5 |
| 40 | 40 ± 3 | 0.060 | 0.039 | 99.3 |
Матричный эффект оценивали как относительное смещение bмэ, сравнивая относительные сигналы аналита, введенного в органический растворитель (Sотн, р) и в образец мочи (Sотн, м) (табл. 9). Расчет осуществляли по следующей формуле:
(2)
${{b}_{{{\text{М}}Э}}} = \left| {{{S}_{{{\text{отн, м}}}}}--{{S}_{{{\text{отн, р}}}}}} \right| \times {{100} \mathord{\left/ {\vphantom {{100} {{{S}_{{{\text{отн, р}}}}}}}} \right. \kern-0em} {{{S}_{{{\text{отн, р}}}}}}}.$Таблица 9.
Матричный эффект при определении 1‑гидроксипирена
| № образца | Количество 1-гидроксипирена, нг |
Sотн, р | Sотн, м | bMЭ, % |
|---|---|---|---|---|
| 1 | 0.4 | 0.00417 | 0.00423 | 1.6 |
| 2 | 4 | 0.0409 | 0.0415 | 1.5 |
| 3 | 20 | 0.219 | 0.210 | 3.9 |
| 4 | 40 | 0.435 | 0.446 | 2.66 |
Максимальный матричный эффект составил 3.9%.
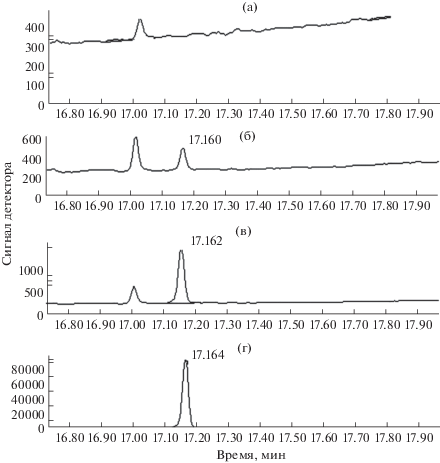
На рис. 1 приведены масс-хроматограммы холостого образца и трех образцов с разными концентрациями 1-гидроксипирена (0.1, 0.5, 39 нг/мл) (рис. 1). Пик триметилсилил-1-гидроксипирена узкий (полуширина пика 1.5 c), симметричный, мешающие пики отсутствуют. Следовательно, достигнута высокая селективность определения. Значение sr измерения времен удерживания составляет 0.027.
Рис. 1.
Масс-хроматограммы (при m/z 290) холостого образца (а), образцов мочи с концентрацией 1-гидроксипирена 0.1 нг/мл (б), 0.5 нг/мл (в), образца мочи работника, подвергшегося воздействию ПАУ (концентрация 1-гидроксипирена 39 нг/мл) (г).
Разработанная методика количественного определения 1-гидроксипирена в моче апробирована на образцах мочи работников (31 человек) производства алюминия. Содержание аналита в моче составляло от 0.1 до 263 нг/мл. Проанализированы также образцы мочи работников других производств (14 человек), где в воздушной среде отсутствуют ПАУ; концентрации 1-гидроксипирена находились в интервале от 0.08 до 0.9 нг/мл.
* * *
Таким образом, применение данной методики позволило снизить продолжительность анализа за счет уменьшения времени ферментативного гидролиза и дериватизации 1-гидроксипирена по сравнению с известными методиками. Также достигнуты высокая точность определения за счет использования изотопно-меченного стандарта 1-гидроксипирена-d9 и повышение чувствительности определения за счет увеличения степени экстракции из биологической матрицы.
Список литературы
Jongeneelen F.J., Boss R.P., Anizon R.B., Theuws J.L., Henderson P.T. Biological moinitoring of polycyclic aromatic hydrocarbons metabolites in urine // Scand. J. Work Environ. Health. 1986. V. 12. P. 137.
Jurgen J., Albrecht S. Biomonitoring of polycyclic aromatic hydrocarbons in human urine // J. Chromatog. B. 2002. V. 778. P. 31.
Rossella F., Campo L., Pavanello S., Kapka L., Sivinska E., Fustinoni S. Urinary polycyclic aromatic hydrocarbons and metabolites as biomarkers of exposure in coke oven workers // Occup. Environ. Med. 2009. V. 66. P. 509.
Jongeneelen F.J., Anzion R.B.M., Henderson P.T. Determination of hydroxylated metabolites of polycyclic aromatic hydrocarbons in urine // J. Chromatog. 1987. V. 413. P. 227.
Campo L., Rossella F., Fustinoni S. Development of a gas chromatography/mass spectrometry method to a quantify several urinary monohydroxy metabolites of polycyclic aromatic hydrocarbons in occupationally exposed subjects // J. Chromatog. B. 2008. V. 875. P. 531.
Ho-Sang Shin, Hyun-Hee Lim. Simultaneous determination of 2-naphtol and 1-hydroxypyrene in urine by gas chromatography-mass spectrometry // J. Chromatog. B. 2011. V. 879. P. 489.
Blau K., Halket J.M. Handbook of Derivates for Chromatography. 2nd ed., New-York: Wiley, 1993. 369 p.
Knapp D.R. Handbook of Analytical Derivatization Reaction. New-York: Wiley, 1979. 741 p.
Pierce A.E. Silylation of Organic Compounds. Rockford, Illinois: Pierce chemical Co., 1968. 487 p.
Schummer C., Delhomme O., Appenzeller B.M.R. Comparison of MTBSTFA and BSTFA in derivatization reaction of polar compounds prior to GC/MS analysis // Talanta. 2009. V. 77. P. 1473.
Смагунова А.Н., Пашкова Г.В., Белых Л.И. Математическое планирование эксперимента в методических исследованиях аналитической химии. Иркутск: Изд-во ИГУ, 2015. 137 с.
Guideline on bioanalytical method validation, Europe medicinal agency. 2011. www.ema.europa.eu/en/documents/scientific-guideline/guideline-bioanalytical-method-validation_en.pdf (21.07.2011).
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии