

Журнал аналитической химии, 2019, T. 74, № 3, стр. 218-224
Об особенностях линии FeLα1,2, используемой в рентгенофлуоресцентном анализе для оценки степени окисления железа
Т. Г. Кузьмина 1, *, М. А. Тронева 1, Т. В. Ромашова 1
1 Институт геохимии и аналитической химии им. В.И. Вернадского Российской академии наук
119991 Москва, ул. Косыгина, 19, Россия
* E-mail: kuzminatg@inbox.ru
Поступила в редакцию 17.01.2017
После доработки 05.09.2018
Принята к публикации 05.09.2018
Аннотация
Исследованы некоторые особенности валентной линии FeLα1,2: показана связь между интенсивностью линии и степенью окисления железа, установлена зависимость интенсивности линии FeLα1,2 от присутствия в пробе других элементов.
Попытки применить рентгенофлуоресцентный анализ (РФА) для определения не только валового содержания элемента в пробе, но и его валентной формы предпринимались с самого начала использования рентгеноспектральных приборов в аналитической химии и продолжаются до сих пор. Однако задача определения элементов с той или иной степенью окисления так до конца и не решена.
В работе [1] подробно рассмотрены различные подходы к решению этой проблемы с использованием современного рентгеновского оборудования. Показано, что стандартные спектрометры с волновой дисперсией, предназначенные для проведения массовых анализов, имеющие большую светосилу и достаточно низкие пределы обнаружения, обладают сравнительно невысоким разрешением. В связи с этим определение степени окисления элемента по величине смещения его эмиссионной линии или изменению ее формы не представляется возможным. Наиболее перспективным, по мнению авторов работы [1], является использование в качестве аналитического сигнала отношения интенсивностей “последней” эмиссионной линии элемента к нормирующей линии, не являющейся валентной, но близкой по своей длине волны к валентной. Это позволяет исключить зависимость этой величины от валового содержания элемента, пренебречь различием в массовых коэффициентах ослабления эмиссионной линии и другими матричными эффектами. Так, для определения степени окисления железа предложено [1] использовать отношение интегральных интенсивностей линий FeKβ2 и FeKβ1 (рис. 1) во втором порядке отражения. Приведены экспериментально полученные значения этого отношения для Fe, FeO и Fe2O3, которые составили 0.051, 0.023 и 0.036 соответственно. Однако о методике определения валентных форм железа речь не идет.
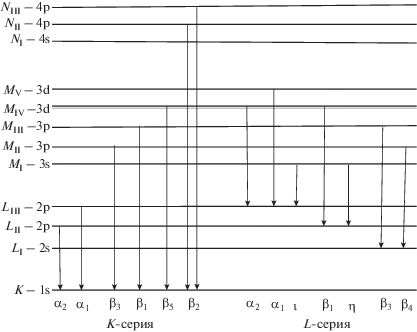
В работах [2, 3] для определения валентного состояния железа в образцах горных пород и железных руд используют отношение интенсивностей, измеренных на максимумах линий FeKβ5 и FeKβ1,3 в первом порядке отражения. Получены регрессионные уравнения, связывающие это отношение с отношением содержаний FeO и Fe2O3общ или с величиной N, характеризующей зарядовое состояние железа и рассчитанное по формуле:
Для горных пород удовлетворительная погрешность градуировки достигается путем разделения их на 10 групп, близких по минеральному и химическому составам. Авторы отмечают, что существуют трудности в определении положения максимума пика валентной линии FeKβ5 и точек замера фона из-за ее малой интенсивности и наложения на линию FeKβ1,3. Это особенно критично для проб с низким содержанием железа.
Кроме того, показано [3], что в качестве аналитического сигнала в РФА возможно использование отношения интенсивностей линий L-серии: валентной линии FeLβ1, соответствующей переходу с уровня MIV на уровень LII, к линии Fe-Lα1,2, соответствующей переходу с уровней MIV,V на уровень LIII. Но из приведенного авторами графика зависимости этого отношения от величины N для образцов оксидных и силикатных соединений железа видно, что погрешность градуировки значительна.
В более ранних исследованиях [4–6] на электронно-зондовых рентгеноспектральных микроанализаторах (РСМА), где характеристические линии переходов на уровни LII и LIII хорошо разрешаются, для определения степени окисления железа также использовали линии Lα1,2 и Lβ1. Так, в работе [5] исследованы LII,III-эмиссионные спектры и отношения интенсивностей линий Lα1,2 и Lβ1 металлического железа и оксидов Fe2O3 и Fe3O4. Показано, что величина этого отношения существенно выше для Fe2O3 и Fe3O4, чем для металла. Авторы работы [4] установили, что обратное отношение ILβ1/ILα1,2 систематически увеличивается с ростом отношения Fe2+/Fe3+. Вместе с тем сделан вывод, что трудно полностью отделить эффект, связанный со степенью окисления элемента, от других эффектов, таких как поглощение и дополнительное возбуждение флуоресцентного излучения внутри образца. В работе [6] также отмечена зависимость отношения интенсивностей линий L-спектра от присутствия в пробе других элементов, но не проведены специальные исследования и не установлены какие-либо закономерности.
Эксперименты, выполненные на рентгенофлуоресцентном спектрометре [3], показали, что интенсивность линии FeLβ1 уменьшается при переходе от двухвалентного железа (FeTiO3) к трехвалентному (Fe2O3, FeOOH, Fe2O3 · nH2O). Так как авторы рассматривали спектры, нормированные на интенсивность линии FeLα1,2, то это соответствует уменьшению отношения ILβ1/ILα1,2, что, в свою очередь, согласуется с выводами, полученными в работах [4, 5].
Следует отметить, что, в отличие от РСМА, серийные рентгенофлуоресцентные спектрометры имеют недостаточное разрешение для разделения линий FeLβ1 и FeLα1,2 (рис. 2), что создает проблему корректного измерения интенсивности линии FeLβ1. Кроме того, ни одна из этих линий не является нормирующей, так как обе они связаны с переходами с валентных уровней MIV и MV.
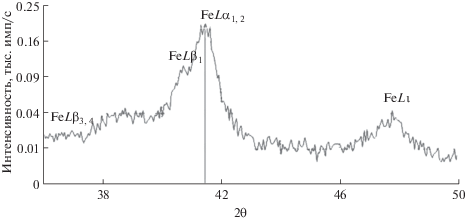
Нами рассмотрена возможность альтернативного подхода к использованию валентной линии FeLα1,2 для оценки степени окисления железа в геологических пробах. Из всех валентных линий железа K- и L-серий именно линия FeLα1,2 (17.59 Å) выражена наиболее четко, хотя имеют место наложения линий FeLβ1 (17.26 Å) и MnLβ3,4 (17.19 Å). В работе экспериментально исследованы особенности этой линии: зависимость ее интенсивности от степени окисления железа и от влияния матричных эффектов – ослабления и возбуждения.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Использовали рентгеновский спектрометр “Axios Advanced” (PANalytical, Holland B.V.), оснащенный рентгеновской трубкой с Rh-анодом и рентгенооптической системой по Соллеру (кристалл PX-1, коллиматор 150 μm). Режим работы рентгеновской трубки 50 кВ/60 мА. Излучение регистрировали газопропорциональным счетчиком. Учитывая наложения линий, интенсивности линии FeLa1,2 и фона измеряли с использованием “тонкого” коллиматора; при этом фон определяли с длинноволновой стороны.
В работе использовали стандартные образцы состава (СОС) горных пород и бинарные смеси FeO или Fe2O3 с оксидами: MgO, Al2O3, SiO2, TiO2, MnO2, CuO.
Излучатели готовили путем прессования в таблетки диаметром 20 мм растертого до 200 меш исходного материала весом 300 мг с добавлением в качестве связующего вещества полистирола (С8Н8) в соотношении 5 : 1.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
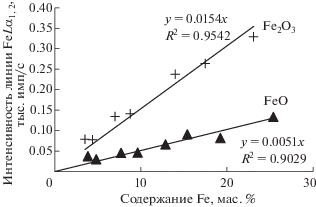
Для изучения особенностей линии FeLα1,2 в присутствии атомов железа (II, III) на данном этапе исследований готовили бинарные пробы, содержащие Fe2+ (FeO) или Fe3+ (Fe2O3), которые разбавляли оксидами Al2O3 и SiO2. Содержание железа в пробах менялось от 0 до ~25 мас. %. Полученные усредненные графики зависимости интенсивности линии FeLα1,2 от содержания железа представлены на рис. 3. Графики линейны с достаточно высокой степенью корреляции, однако углы наклона для железа с разной степенью окисления различны.
Рис. 3.
Зависимость интенсивности линии FeLα1,2 от содержания железа, входящего в состав FeO или Fe2O3 (в качестве наполнителей использованы SiO2 и Al2O3).
Суммарную интенсивность линии FeLα1,2 в пробе, содержащей как FeO, так и Fe2O3 можно представить также в виде линейного уравнения:
(1)
${{I}_{{L\alpha 1,2}}} = A + Bc({\text{FeO}}){\text{ }} + D{\text{с }}({\text{F}}{{{\text{e}}}_{2}}{{{\text{O}}}_{3}}).$Здесь с(FeO) и с(Fe2O3) – содержания оксидов железа, мас. %; A, B, D − константы уравнения, которые могут быть определены методом наименьших квадратов с помощью стандартных образцов состава.
Мы использовали 30 СОС ультраосновных, основных, средних и кислых горных пород (табл. 1). Получено уравнение регрессии:
(2)
$IL{{\alpha }_{{1,2}}} = {\text{ }}0.00554c({\text{FeO}}){\text{ }} + {\text{ }}0.00713c({\text{F}}{{{\text{e}}}_{2}}{{{\text{O}}}_{3}}).$Таблица 1.
Диапазоны содержания оксидов в стандартах, используемых при градуировке
| Оксид | Диапазон содержания, мас. % | Оксид | Диапазон содержания, мас. % |
|---|---|---|---|
| SiO2 | 25−72 | CaO | 0.51−17 |
| Al2O3 | 0.74−22 | Na2O | 0.03−4.4 |
| Fe2O3 | 0.75−10.2 | K2O | 0.005−8.75 |
| FeO | 0.2−14 | TiO2 | 0.05−6.75 |
| Fe2O3общ | 0.5−21 | MnO | 0.04−0.5 |
| MgO | 0.21−43 | Потери при прокаливании | 0.3−25.0 |
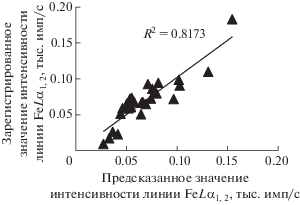
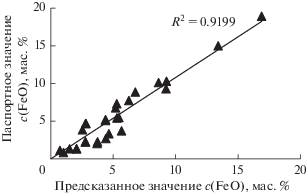
На рис. 4 показана корреляция между значениями интенсивностей линии FeLα1,2, измеренными на стандартных образцах и рассчитанными по уравнению (2).
Рис. 4.
Соотношение между зарегистрированными и предсказанными значениями интенсивности линии FeLα1,2 в стандартных образцах состава, используемых для градуировки.
Из уравнения (1) можно получить уравнение регрессии для расчета содержания оксида железа(II):
(3)
$c{\text{(FeO)}} = K + MIL{{\alpha }_{{1,2}}} + Nc\left( {{\text{F}}{{{\text{e}}}_{2}}{{{\text{O}}}_{3}}} \right),$Учитывая, что с(Fe2O3) = с(Fe2O3)общ– 1.1с(FeO), уравнение (3) можно представить в виде:
(4)
$c({\text{FeO)}} = K{\text{' + }}M{\text{'}}IL{{\alpha }_{{1,2}}} + N{\text{'}}c{{\left( {{\text{F}}{{{\text{e}}}_{{\text{2}}}}{{{\text{O}}}_{{\text{3}}}}} \right)}_{{{\text{о б щ }}}}}.$Значение с(Fe2O3)общ можно получить из РФА валового состава пробы. На рис. 5 показано соотношение между полученными и паспортными значениями с(FeO) в СОC, используемых для градуировки. Также наблюдается заметная корреляция. Однако погрешность градуировки, представленная в виде относительтного стандартного отклонения, значительна и составляет порядка 20%. Полагая, что это может быть обусловлено влиянием матричного эффекта, связанного с ослаблением флуоресцентного излучения линии FeLα1,2 (17.59 Ǻ), мы рассчитали массовые коэффициенты ослабления этой линии (μLα) макроэлементами пробы с Z < 26. Использовали формулу:
Рис. 5.
Соотношение между паспортными и полученными значениями содержания FeO в стандартных образцах состава, используемых для градуировки.
Коэффициенты С, α, β взяты из работы [7] (табл. 2).
Таблица 2.
Коэффициенты С, α и β для вычисления массовых коэффициентов ослабления излучения с длиной волны λLα = 17.59 Å макрокомпонентами пробы
| Z | Длины волн | C | α | β |
|---|---|---|---|---|
| 3 ≤ Z ≤ 9 | λК > λLα | 5.40 × 10–3 | 2.92 | 3.07 |
| 10 ≤ Z ≤ 18 | λK < λLα < λLI | 5.33 × 10–4 | 2.74 | 3.03 |
| 19 ≤ Z ≤ 23 | λK < λLα < λLI | 9.59 × 10–4 | 2.7 | 2.9 |
| Z = 25 | λLI < λLα < λLII | 3.56 | 2.69 | 0 |
| Z = 26 | λLα > λLIII | 2.73 × 10–5 | 2.44 | 3.47 |
Внесение поправки на ослабление флуоресцентного излучения в уравнение (1) приводит его к виду (поправкой на поглощение первичного излучения пренебрегаем):
(6)
$IL{{\alpha }_{{1,2}}}{{\mu }_{{L\alpha }}} = A{\text{'}} + B{\text{'}}c({\text{FeO}}) + D{\text{'}}c({\text{F}}{{{\text{e}}}_{{\text{2}}}}{{{\text{O}}}_{3}}).$Однако введение этой поправки не привело к улучшению результатов. Более того, вопреки ожиданию при одном и том же содержании железа интенсивность флуоресцентной линии FeLα1,2 оказалась выше в тех пробах, в которых коэффициент поглощения имел более высокие значения.
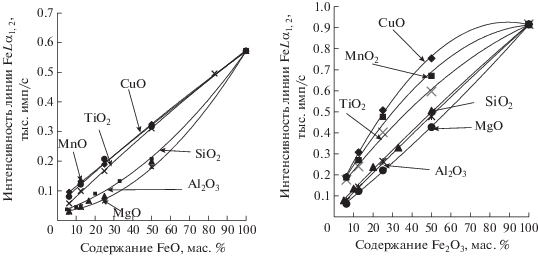
Для более детального исследования полученного эффекта использовали серии бинарных смесей, содержащих FeO или Fe2O3 (6.25, 12.5, 25, 50, 100%) с добавлением одного из оксидов: MgO, Al2O3, SiO2, TiO2, MnO2, CuO. Из рис. 6 видно, что как для FeO, так и для Fe2O3 интенсивности линии FeLα1,2 тем больше, чем тяжелее наполнитель бинарной смеси. Это подтверждает результаты предыдущего эксперимента.
Рис. 6.
Зависимость интенсивности линии FeLα1,2 от содержания оксида железа в присутствии разных наполнителей.
Для того чтобы исключить зависимость аналитического сигнала от влияния матричного эффекта, представлялось логичным использовать отношение интенсивностей линий FeLα1,2 и FeLι. Линия FeLι четко выражена на спектрограмме (рис. 2), соответствует переходу с более глубокого уровня MI на уровень LIII и имеет точно такие же условия возбуждения, как и линия FeLα1. Это подтверждается и полученными значениями интенсивностей линии FeLι, зарегистрированными для бинарных смесей Fe2O3 с MgO, Al2O3, TiO2 и MnO2 в соотношении 1 : 1:
| Оксиды | MgO + + Fe2O3 | Al2O3 + + Fe2O3 | TiO2 + + Fe2O3 | MnO2 + + Fe2O3 | FeO, 100% | Fe2O3, 100% |
| Интенсивность линии Lι, имп/с | 56.2 | 76.1 | 96.6 | 107.7 | 116 | 158 |
Интенсивность линии Lι, так же как и линии Lα1,2, увеличивается с увеличением среднего атомного номера наполнителя. Однако, как показал эксперимент, эта линия не удовлетворяет критерию нормирующей линии. Ее интенсивность не пропорциональна содержанию Fe2O3общ, а меняется в зависимости от степени окисления элемента так же, как и в случае внешних валентных линий (см. выше).
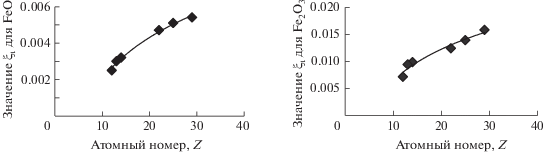
Мы попробовали в достаточно грубом приближении учесть особенности возбуждения линии FeLα1,2 элементами, определяющими макросостав пробы. С этой целью параболы, полученные для бинарных смесей FeO и Fe2O3 c оксидами MgO, Al2O3, SiO2, TiO2, MnO2, CuO (рис. 6), аппроксимировали отрезками прямых линий, проходящими через начало координат, в диапазоне содержания оксидов железа от 0 до 30%. В качестве условного параметра использовали значения тангенсов угла наклона (tg θ = ξ) графиков зависимости интенсивности линии от содержания FeO (ξ2i) и Fe2O3 (ξ3i) в присутствии того или иного наполнителя (i). Для оксидов-наполнителей тех макрокомпонентов, данные по которым отсутствовали, значение ξ как для двух-, так и для трехвалентного железа получали путем интерполяции из эмпирических зависимостей ξ = f(Z) (здесь Z – атомный номер элемента, оксид которого использован в бинарной смеси), представленных на рис. 7. Неясным остался вопрос о том, как возбуждается двухвалентное железо в присутствии трехвалентного и наоборот. Весьма приблизительные оценки этих параметров также получили из зависимостей ξ = f(Z). Рассчитали средние значения тангенсов угла наклона как для двух-, так и для трехвалентного железа (ξ2, ξ3) для всех СОС, используемых в градуировке, с учетом состава их матриц по формулам:
(7)
${{\xi }_{2}} = \frac{{\sum\limits_{\text{i}}^n {{{{\xi }}_{{2{\text{i}}}}}{{с }_{{\text{i}}}}} }}{{\sum\limits_{\text{i}}^n {{{с }_{{\text{i}}}}} }},\,\,\,\,{{\xi }_{3}} = \frac{{\sum\limits_{\text{i}}^n {{{{\xi }}_{{3{\text{i}}}}}} {{с }_{{\text{i}}}}}}{{\sum\limits_{\text{i}}^n {{{с }_{{\text{i}}}}} }},$Рис. 7.
Зависимость значения ξ от порядкового номера элемента, оксид которого добавлен к FeO или Fe2O3.
Далее рассчитали средние значения ξср для каждой пробы по формуле:
(8)
${{\xi }_{{{\text{с р }}}}} = \frac{{{\;}{{{\xi }}_{2}}с \left( {{\text{FeO}}} \right)\quad + {\;}{{{\xi }}_{3}}с \left( {{\text{F}}{{{\text{e}}}_{2}}{{{\text{O}}}_{3}}} \right)}}{{с \left( {{\text{FeO}}} \right)\quad + \quadс \left( {{\text{F}}{{{\text{e}}}_{2}}{{{\text{O}}}_{3}}} \right)}}.$Поправку в уравнение (4) вносили следующим образом:
(9)
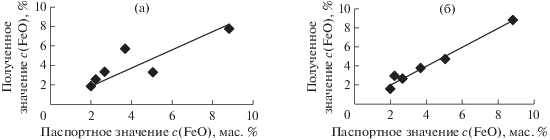
$c{\text{(FeO)}} = G + \frac{{RIL{\alpha }1,2}}{{{{{\xi }}_{{{\text{cр }}}}}}} + Qc{{\left( {{\text{F}}{{{\text{e}}}_{2}}{{{\text{O}}}_{3}}} \right)}_{{{\text{о б щ }}}}},$Кроме того, СОС разделили на группы по их валовому химическому составу: кислые, ультраосновные, основные и средние и получили регрессионные уравнения (9) для каждой группы. Из табл. 3 видно, что при введении поправки по сравнению с первоначальным уравнением относительное стандартное отклонение, характеризующее погрешность градуировки, уменьшается для основных, средних и ультраосновных пород в 1.5–2 раза. Для кислых пород уравнения без поправок и с поправками имеют равнозначные результаты. В качестве примера на рис. 8 приведены графики зависимости полученных содержаний FeO от паспортных значений для СОС ультраосновных пород, используемых в градуировке.
Таблица 3.
Относительные погрешности градуировки (sr, %) при использовании уравнений (4) и (9)
| Породы | Уравнение (4) | Уравнение (9) |
|---|---|---|
| Основные и средние | 16 | 10 |
| Кислые | 12 | 11 |
| Ультраосновные | 30 | 15 |
Рис. 8.
Расчетные и паспортные значения содержаний FeO в стандартных образцах состава ультраосновных пород: (a) – без поправки по уравнению (4), (б) – с использованием поправки по уравнению (9).
Таким образом, интенсивность валентной линии FeLα1,2 рентгеновского спектра увеличивается при переходе от Fe2+ к Fe3+, т.е. концентрационная чувствительность различна для железа с разной степенью окисления. По-видимому, это является характерной особенностью валентных линий железа. Более того, даже, казалось бы, невалентная линия Lι имеет такую же особенность, что является артефактом и требует теоретического обоснования. Для линии Lβ1 (нами не измерена из-за ее низкой интенсивности), используемой в работах [2–5], также должна иметь место подобная зависимость. Однако коэффициенты, характеризующие чувствительность определения содержаний Fe2+ или Fe3+ по разным валентным линиям K- и L-спектров (Kβ2, Kβ5, Lα1,2, Lβ1, Lι), вероятнее всего, будут различаться. Поэтому авторам работ [2–5] не удалось получить удовлетворительные результаты при оценке степени окисления железа в пробах различного состава, используя зависимость отношения ILβ1/ILα1,2 от с(FeO)/с(Fe2O3). Замена с(Fe2O3) на с(Fe2O3)общ также некорректна, так как в этом случае необходимо использовать нормирующую линию, интенсивность которой была бы пропорциональна содержанию Fe2O3общ. С валентными линиями это условие не выполняется. Более того, по-видимому, в L-спектре отсутствует линия, которую можно было бы использовать в качестве нормирующей.
Другой отличительной чертой линии FeLα1,2 является особенность зависимости ее интенсивности от присутствия в пробе других элементов. Интенсивность увеличивается с увеличением коэффициента ослабления (т.е. среднего атомного номера наполнителя). Этот эффект объясняется так же, как и возбуждение флуоресценции элементов с малыми атомными номерами (Z), так как край поглощения линии FeLα − LIII (17.52 Å) располагается в длинноволновой области спектра.
Показано [8], что при ионизации K-оболочек элементов с малыми Z роль первичных фотонов (из-за поглощения длинноволнового излучения в окне рентгеновской трубки) не является определяющей. Значимыми оказываются такие процессы ионизации, как избирательное возбуждение атомов за счет L-излучения элементов матрицы, возникающего в результате внутриатомных каскадных переходов, а также возбуждение фото- и Оже-электронами элементов со средними и высокими атомными номерами. Для линии FeLα1,2 влияние матрицы определяется также эффектами возбуждения, которые оказываются тем значительней, чем больше средний атомный номер наполнителя.
Очевидно, что даже приближенный учет влияния матричного состава пробы на возбуждение линии FeLα1,2 позволяет существенно улучшить качество градуировки. Однако необходимы более точные поправки или поиск вариантов косвенного учета особенностей возбуждения уровня LIII.
* * *
Таким образом, удалось разделить два эффекта, влияющих на интенсивность валентной линии FeLα1,2: эффект валентности и матричный эффект. Показано, что из-за различия в значениях концентрационной чувствительности для атомов железа с разной степенью окисления в качестве функции отклика можно использовать не только отношение интенсивностей линий (валентной к нормирующей), но и интенсивность одной линии FeLα1,2. Учет особенностей возбуждения линии Lα приводит к снижению погрешности градуировки и повышению точности анализа.
Список литературы
Филиппов М.Н., Куприянова Т.А., Лямина О.И. Одновременное определение содержания и формы нахождения элемента в твердом теле рентгенофлуоресцентным методом // Журн. аналит. химии. 2001. Т. 56. № 8. С. 817. (Filippov M.P., Kupriyanova T.A., Lyamina O.I. Simultaneous determination of the concentration of elements and their speciation in solid samples using X-ray fluorescence spectrometry // J. Analyt. Chem. 2001. V. 56. № 8. P. 729.)
Чубаров В.М., Финкельштейн А.Л. Рентгенофлуоресцентное определение отношения FeO/Fe2O3общ в горных породах // Журн. аналит. химии. 2010. Т. 65. № 6. С. 634. (Chubarov V.M., Finkel’shtein A.L. X-ray fluorescence determination of the FeO/Fe2O3tot ratio in rocks// J. Analyt. Chem. 2010. V. 65. № 6. P. 620.)
Чубаров В.М., Финкельштейн А.Л., Суворова Л.Ф., Костровицкий С.И. Определение валентного состояния железа в пикроильмените методами рентгеновского электронно-зондового микроанализа и рентгенофлуоресцентного анализа // Записки рос. минерал. общества. 2012. Ч. CXLI. № 2. С. 83.
Albee A.L., Chodos A.A. Semiquantitative electron microprobe determination of Fe2+/Fe3+ and Mn2+/Mn3+ in oxides and silicates and its application to petrologic problems // Am. Mineralogist. 1970. V. 55. № 3–4. P. 491.
Fischer D.W. Changes in the soft X-ray L-emission spectra with oxidation of the first series transition metals // J. Appl. Phys. 1965. V. 36. № 6. P. 2048.
O`Nions R.K., Smith D.G. Investigations of the LII,III X‑ray emission spectra of Fe by electron microprobe. Part 2. The Fe LII,III spectra of Fe and Fe–Ti oxides // Am. Mineralogist. 1971. V. 56. P. 1452.
Верховодов П.А. Рентгеноспектральный анализ. Вопросы теории и способы унификации. Киев: Наукова Думка, 1984. 160 с.
Павлинский Г.В. Основы физики рентгеновского излучения. М.: ФИЗМАТЛИТ, 2007. 240 с.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии