

Журнал аналитической химии, 2022, T. 77, № 8, стр. 755-762
Мицеллярная тонкослойная хроматография и концентрирование L-карнитина наночастицами магнетита
Е. Г. Сумина a, *, С. Н. Штыков a, **, И. С. Решетникова a, Н. А Юрасов a, В. З. Угланова a
a Саратовский национальный исследовательский государственный университет
имени Н.Г. Чернышевского, Институт химии
410012 Саратов,
ул. Астраханская, 83, Россия
* E-mail: suminaeg@yandex.ru
** E-mail: shtykovsn@mail.ru
Поступила в редакцию 08.07.2021
После доработки 19.11.2021
Принята к публикации 22.11.2021
- EDN: FDGNJA
- DOI: 10.31857/S0044450222080163
Аннотация
Методом тонкослойной хроматографии (ТСХ) изучено хроматографическое поведение L-карнитина в водно-органических подвижных фазах (ПФ) и водной ПФ в присутствии катионного ПАВ хлорида цетилпиридиния (ЦПХ) на пластинках с нормальной неподвижной фазой на основе силикагеля. Среди водно-органических ПФ лучшие результаты получены для ПФ состава пропанол-2–ацетон–вода–уксусная кислота (4 : 5 : 2 : 2). Наилучшие хроматографические характеристики получены для водно-мицеллярной ПФ, содержащей 0.001 М ЦПХ. Впервые изучена сорбция L-карнитина на наночастицах магнетита, модифицированных бромидом цетилтриметиламмония. Установлено, что количественная сорбция аналита со степенью извлечения L-карнитина 89% протекает за 40 мин. Найден элюент – этанол, позволяющий на 95% десорбировать L-карнитин. Установлено, что применение для концентрирования 3 мг наномагнетита позволяет в пять раз уменьшить нижнюю границу определяемых содержаний L-карнитина. Разработана методика денситометрического определения L-карнитина с помощью мицеллярной ТСХ в напитке для спорта XXI Power “L-КАРНИТИН”.
L-карнитин – природное вещество, синтезируемое организмом, основная роль которого состоит в переносе длинноцепочечных жирных кислот в митохондрии клеток через их внутреннюю мембрану для последующего окисления и получения энергии [1–3]. L-карнитин обладает также ярко выраженным комплексом антиокислительных и антимутагенных свойств, антиангинальным и противоишемическим эффектами, участвует в регуляции энергетического обмена сердца и вследствие этого широко используется в медицине [4–6], в том числе для поддержки здоровья пожилых людей [7] и развития выносливости в спорте [8]. Известной проблемой является дефицит карнитина у детей до 15 лет [9]. Карнитин и его производные содержатся в мясных продуктах, молоке, яйцах, рыбе, орехах, авокадо и многих других продуктах, широко применяется в биологически активных добавках (БАД), энергетических напитках [10]. Все это требует разработки разнообразных методов его определения в биологических объектах (тканях, жидкостях), продуктах питания, пищевых добавках, БАД и фармпрепаратах.
Анализ методов определения карнитина дан в обзорах [11–14]. Показано, что карнитин определяют в нескольких формах: в свободном состоянии, в виде эфиров с короткой, средней и длинной алкильными цепочками (С1–С18), а также его суммарное содержание. Эфиры карнитина различают по их растворимости в воде. В связи с этим рассматриваются методы выделения и разделения L-карнитина и его ацильных производных [15, 16].
Вследствие сложного состава, нелетучести и необходимости определения как отдельных составляющих, так и суммарного содержания карнитина наиболее часто для его определения применяют метод обращенно-фазовой высокоэффективной жидкостной хроматографии [11–15] с разными вариантами детектирования: спектрофотометрическим [17], флуориметрическим [18, 19] детекторами после реакции дериватизации, а также кондуктометрическим [20], масс-спектрометрическим [21] и др. Кроме этого, карнитин определяют капиллярным электрофорезом [22], ионной хроматографией [23], проточно-инжекционным методом анализа [24] и различными вариантами масс-спектрометрии [25]. Так как карнитин не содержит хромофорных групп, то его фотометрическое [26], флуориметрическое [27] и радиоферментное [10], и даже хроматографическое определение [18] основано на предварительной дериватизации при взаимодействии с различными ферментами [15]. Важное значение имеет возможность раздельного определения энантиомеров карнитина, поскольку содержание D-изомера строго нормируется [19]. Для выделения карнитина из объектов и концентрирования обычно используют жидкостно-жидкостную экстракцию и микроэкстракцию или твердофазную экстракцию (ТФЭ) [15, 16, 18, 28, 29].
Одним из вариантов ТФЭ является магнитная твердофазная экстракция (МТФЭ), основанная на использовании для извлечения аналита коллоидного раствора наночастиц магнетита, обладающих суперпарамагнитными свойствами. Это явление позволяет быстро в течение нескольких десятков секунд отделять сорбент с аналитом от жидкой матрицы с помощью внешнего магнитного поля [30–34]. Сорбционное концентрирование L-карнитина с использованием магнитных наночастиц не описано.
Анализ литературы показал, что определению карнитина методом тонкослойной хроматографии (ТСХ) посвящена только одна работа, в которой описан анализ пищевых добавок, напитков, капсул и таблеток на содержание карнитина [35]. Неподвижной фазой являлись целлюлозные пластины для высокоэффективной ТСХ, а подвижной фазой − смесь метанол–вода–ледяная уксусная кислота (10 : 2 : 0.1, по объему). Хроматограммы анализировали денситометрически при 420 нм после обработки нингидрином.
Более 25 лет назад широкое применение в качестве подвижных фаз в ТСХ нашли мицеллярные подвижные фазы (МПФ), исключающие применение летучих, канцерогенных и токсичных растворителей. Показано, что метод мицеллярной ТСХ (МТСХ) не уступает, а часто превосходит по эффективности традиционный вариант ТСХ с органическими элюентами [36–40]. Метод МТСХ применяли и для определения аминокислот, к классу которых относится карнитин, но поведение карнитина не исследовали [37–39].
Цель настоящей работы − применение МПФ для мицеллярной тонкослойной хроматографии L-карнитина на пластинках с прямой фазой, а также применение концентрирования методом МТФЭ на наночастицах магнетита для снижения нижней границы определяемых содержаний.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Реагенты. Использовали L-карнитин (Sigma Aldrich, США) с содержанием основного вещества более 95%. Стандартный водный раствор L-карнитина (L-Кр) с концентрацией 20 мг/мл готовили по точной навеске. Магнетит синтезировали в деионизованной воде смешиванием в соотношении 1 : 2 растворов FeCl2⋅4H2O и FeCl3⋅6H2O (99.8%, Acros Organics, Германия) с гидроксидом натрия (99.8%, Fluka, Германия) [32, 41]. Для создания инертной среды через раствор пропускали азот (ГОСТ 9293-74). Магнетит стабилизировали и модифицировали катионным поверхностно-активным веществом (ПАВ) – бромидом цетилтриметиламмония (ЦТАБ) (99%, Acros Organics, Германия), по методике [41]. Использовали 0.06 М водный раствор ЦТАБ. Ацетатно-аммиачные буферные растворы (ААБ) (рН 3–8) готовили смешиванием необходимых количеств 2 М растворов уксусной кислоты и аммиака. Ионную силу раствора в водных МПФ создавали раствором хлорида натрия. Для приготовления подвижных фаз (ПФ) использовали водно-органические растворители: пропанол-1 (ч.д.а.), пропанол-2 (ч.д.а.), ацетон (ч.д.а.), уксусную кислоту (ч.д.а.) и мицеллярные растворы хлорида цетилпиридиния (ЦПХ, 96%, НПО “Синтез ПАВ”, Россия). Исходные растворы ЦПХ с концентрацией 0.1 М готовили по точной навеске растворением в дистиллированной воде при нагревании до 40–50°С. Рабочие растворы всех веществ готовили разбавлением исходных перед употреблением.
Аппаратура. Деионизованную воду получали на установке водоочистки УВОИ–“М-Ф” (Медиана-Фильтр, Россия), механическое перемешивание растворов осуществляли на орбитальном шейкере с аналоговым управлением ELMI S-3.02 (ЭЛМИ, Латвия) и с помощью магнитной мешалки MINI MR STANDARD (IKA WERKE, Германия). Для магнитной сепарации наночастиц магнетита использовали постоянный магнит Nd–Fe–B с (ВН)макс = 40 МГсЭ. Дзета-потенциал частиц гидрозоля в растворах магнетита измеряли на анализаторе Zetasizer Nano-Z (Malvern Instruments Ltd, Великобритания). Размеры наночастиц магнетита и толщину покрытия ЦТАБ определяли методом просвечивающей электронной микроскопии (ПЭМ) с использованием просвечивающего электронного микроскопа Libra 120 (Carl Zeiss, Германия) при ускоряющем напряжении 120 кВ.
L-Карнитин определяли на денситометре (Сорбполимер, Краснодар, Россия) с УФ-лампой (TUV PL-S PHILIPS, 254 нм) методом восходящей ТСХ на коммерческих пластинах с нормальной фазой Сорбфил (силикагель ПТСХ-П-А-УФ, ТУ26-11-17-89, размер зерен 5–17 мкм, связующее силиказоль) на полимерной подложке (Сорбполимер, Краснодар, Россия). Объем наносимой пробы составлял 1 мкл. Зону основного вещества L-Кр идентифицировали, обрабатывая пластину свежеприготовленным 0.1 н раствором перманганата калия. После хроматографирования и проявления пластинки сушили в течение 10 мин при 90°С, охлаждали на воздухе, рассчитывали значения Rf аминокислоты, а также параметры эффективности разделения (число теоретических тарелок (N) и эффективную высоту теоретической тарелки, ВЭТТ (H)). Площади хроматографических зон измеряли с помощью графического редактора “Adobe Photoshop CC 2018”. Для построения градуировочных графиков использовали исходные растворы L-карнитина (20 мг/мл), из которого готовили рабочие растворы в интервале концентраций от 0.4 до 2.0 мг/мл последовательным разбавлением исходного раствора в объеме 5 мл, отбирая точные аликвоты с помощью дозатора.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Тонкослойная хроматография L-карнитина. Особенность МПФ состоит в том, что в гомогенной водной дисперсной среде дифильные молекулы ПАВ при определенной концентрации (критической концентрации мицеллообразования, ККМ) спонтанно образуют наноразмерные (радиус 1.5–2 нм) динамические наноагрегаты, называемые мицеллами, состоящие из нескольких десятков ионов (молекул) ПАВ. Присутствие в подвижной фазе мицелл улучшает селективность хроматографирования за счет возникновения нового равновесия, в котором сорбат распределяется между водной средой и мицеллярной псевдофазой, дополняющего равновесие в системе ПФ–модифицированный сорбент [36, 42]. Характерным свойством ПАВ, образующих МПФ, является способность изменять полярность поверхности пластин в ТСХ, позволяя в динамическом режиме её гидрофобизировать и превращать нормальную фазу в обращенную, а при избытке ПАВ придавать поверхности заряд и реализовывать ион-парный вариант ТСХ [40].
Хроматографическое поведение L-Кр методом ТСХ изучали с использованием как традиционной водно-органической ПФ, так и мицеллярной ПФ на основе катионного ПАВ хлорида цетилпиридиния. Анализ литературы показал, что обязательными компонентами водно-органических ПФ являются спирт, уксусная кислота, вода, иногда добавляют апротонный растворитель, в качестве которого применяли ацетон. Сравнивали составы пропанол-2–ацетон–вода–уксусная кислота (4 : 5 : 2 : 2), пропанол–ацетон–вода–уксусная кислота (4 : 5 : 2 : 2) и этанол–ацетон–вода−уксусная кислота (4 : 5 : 2 : 2). Установлено, что наилучшие хроматографические характеристики обеспечивает первый вариант ПФ (Rf = = 0.57, N = 454, H = 0.0154).
Ранее на примере других аминокислот показано, что МПФ на основе ЦПХ по сравнению с другими ПАВ позволяют получать наибольшие величины числа теоретических тарелок [43]. При выборе оптимального состава МПФ концентрацию ЦПХ варьировали в интервале 4 × 10–5−4 × 10–2 М, а ионная сила раствора была постоянной и согласно данным [43] составляла 0.05. Увеличение концентрации электролита в растворе до указанной величины приводит к росту подвижности за счет “высаливания” (солюбилизации) аминокислоты из водной среды в мицеллярную псевдофазу [39, 43]. Наилучшие хроматографические характеристики карнитина наблюдались при концентрации ЦПХ 8 × 10–4–1 × 10–3 М (Rf = 0.44, N = = 1708, H = 0.0041). Градуировочные графики для определения карнитина в водно-органической и мицеллярной ПФ и их характеристики приведены на рис. 1 и в табл. 1. Видно, что большей чувствительностью и лучшими метрологическими характеристиками характеризуется определение с использованием мицеллярной ПФ, которую в дальнейшем использовали для определения карнитина в объекте.
Рис. 1.
Градуировочные графики зависимости площади хроматографической зоны L-карнитина от его концентрации в растворе. Подвижная фаза: 1 – водно-органическая, 2 – водно-мицеллярная (без концентрирования). Ионная сила 0.05.
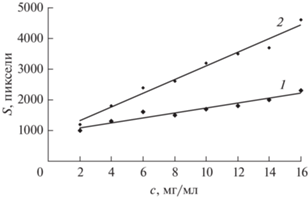
Таблица 1.
Характеристики градуировочных графиков для определения L-карнитина
| Характеристика | Водно-органическая ПФ | МПФ | |
|---|---|---|---|
| без концентрирования | с концентрированием | ||
| Уравнение градуировочного графика | y = 79.8x + 932 | y = 221x + 882 | y = 1544x + 597 |
| R2 | 0.975 | 0.988 | 0.991 |
| Интервал линейности, мг/мл | 2.0–16.0 | 2.0–16.0 | 0.4–1.9 |
Сорбция L-карнитина на магнитных наночастицах (МНЧ), модифицированных бромидом цетилтриметиламмония. Наночастицы магнетита синтезировали в диапазоне рН 9–11 в соответствии с химической реакцией ${\text{F}}{{{\text{e}}}^{{2 + }}} + 2{\text{F}}{{{\text{e}}}^{{3 + }}} + \,\,{\text{8O}}{{{\text{H}}}^{ - }}$ = = ${\text{F}}{{{\text{e}}}_{3}}{{{\text{O}}}_{4}} \downarrow + \,\,4{{{\text{H}}}_{2}}{\text{O}}$ и модифицировали катионами ЦТАБ по разработанной ранее методике [32, 41]. Согласно ПЭМ наночастицы с оболочкой ЦТАБ имеют размер (8 ± 2) нм с толщиной оболочки (1.0 ± 0.1) нм [41]. ζ-Потенциал полученных наночастиц максимален при рН 3–5 [41] и не меняет своего значения до ионной силы, равной 0.1 (NaCl). Это свидетельствует о наибольшей устойчивости коллоидного раствора МНЧ в этой области рН. Можно полагать, что и сорбция L-Кр также будет максимальна в этих условиях.
Эффективность сорбции изучали, определяя методом ТСХ содержание L-Кр в надосадочной жидкости после отделения наночастиц Fe3O4 постоянным магнитом. Значение степени извлечения (RC, %) рассчитывали по формуле (1):
где с0 – молярная концентрация L-Кр в исходном растворе до сорбции; с – концентрация в надосадочной жидкости после сорбции, рассчитанная по уравнению градуировочного графика.
Степень десорбции (RД, %) рассчитывали из отношения количества десорбированного L-Кр к количеству сорбированного наночастицами магнетита по формуле (2):
где сэл – концентрация L-Кр в объеме элюента после десорбции, мкмоль; ссорб – концентрация L-Кр, сорбированного на МНЧ, мкмоль. Сорбцию изучали из объема коллоидного раствора магнетита 5 мл, содержащего магнетит, буферный раствор и аликвоту раствора карнитина.Известно, что на сорбцию и концентрирование веществ на МНЧ влияют различные факторы: рН среды, масса сорбента, время сорбции, а также время и способ перемешивания раствора, которые варьировали при выборе оптимальных условий сорбции. Результаты представлены на рис. 2. Установили, что максимальное значение RC L-Кр, равное 89–90%, достигается при рН 4.5–5.0. Сопоставляя этот интервал рН с величиной рК = 4.2 L-карнитина [44] и с величиной рН изоэлектрической точки 5.2–5.4 магнетита, можно констатировать, что сорбция карнитина обусловлена электростатическим взаимодействием диссоциированной в этой области рН карбоксильной группы аналита и положительно заряженной за счет присутствия катиона цетилтриметиламмония поверхности МНЧ. Уменьшение сорбции L-Кр при рН > 5 может быть связано с резким уменьшением величины положительного ζ-потенциала поверхности наносорбента, который при рН 6 близок к нулю, а при рН 7 имеет уже отрицательное значение [41].
Рис. 2.
Зависимость степени извлечения L-карнитина наночастицами F3O4@ЦТАБ от рН среды (а), массы сорбента (б), времени механического перемешивания (в). Vр-ра = 5 мл, сL-Кр = 4 мг/мл, n = 3, P = 0.95.

Из рис. 2 также видно, что при рН 4.9 для количественного извлечения L-Кр при его концентрации в растворе 4 мг/мл достаточно 3 мг магнетита. Установлено, что сорбционное равновесие достигается за 30–40 мин при степени извлечения 89%. Представленные на рис. 2 условия максимальной сорбции L-Кр модифицированными наночастицами магнетита использовали в дальнейшей работе.
Десорбция L-карнитина с поверхности магнитных наночастиц, модифицированных бромидом цетилтриметиламмония. При выборе растворителя для десорбции апробировали этанол, ацетонитрил, уксусную кислоту, варьируя их объемы от 0.5 до 5 мл. Установили (рис. 3), что количественная десорбция (RД = 95%) L-Кр достигается при использовании 1 мл этанола при механическом перемешивании в течение 20 мин. Применение бóльших объемов этанола нецелесообразно, так как нивелирует преимущества концентрирования.
Рис. 3.
Зависимость степени десорбции L-карнитина от объема этанола (а) и времени перемешивания (б). Ацетатно-аммиачный буферный раствор (рН 4.9), механическое перемешивание, m(F3O4@ЦТАБ) = = 3 мг, Vр-ра = 5 мл, сL-Кр = 4 мг/мл, n = 3, P = 0.95.

На основании сравнения интервалов линейности градуировочных графиков ТСХ-определения L-карнитина без концентрирования и при концентрировании из 5 мл исходного водного раствора установили, что применение концентрирования позволило в 5 раз уменьшить нижнюю границу определяемых содержаний L-Кр с 2.0 до 0.4 мг/мл (табл. 1).
Определение содержания L-карнитина в напитке. Содержание L-карнитина методом мицеллярной ТСХ определяли в напитке для спортсменов XXI Power “L-КАРНИТИН” (Россия). Согласно заявленным данным концентрация карнитина в напитке должна составлять 2.4 мг/мл. Напиток был прозрачным, использовали его для анализа без разбавления. Концентрирование на МНЧ не применяли из-за высокой исходной концентрации L-карнитина. С помощью микродозатора отбирали аликвоты раствора объемом 1 мкл и наносили на стартовую линию хроматографической пластины, выполняя три параллельных определения.
Хроматографический анализ выполняли без предварительного насыщения камеры мицеллярной подвижной фазой согласно рекомендациям [39, 43]. После хроматографирования пластину вынимали из камеры, сушили на воздухе в течение 3–5 мин, затем в сушильном шкафу при 90–100°С до полного удаления растворителя. Далее пластину опрыскивали свежеприготовленным раствором перманганата калия, снова помещали в сушильный шкаф на 5 мин до проявления зон желтого цвета на сиреневом фоне пластины (рис. 4).
Рис. 4.
Хроматограммы L-карнитина в стандартном растворе (1) и исследуемом объекте XXI Power “L-КАРНИТИН” (2). Неподвижная фаза: Сорбфил. По-движная фаза: хлорид цетилпиридиния (1.0 × 10–3 М)–вода, 0.05 М NaCl.

Установили, что величины подвижности L-карнитина в стандартном растворе и в анализируемом напитке совпадают (рис. 2), что явилось основой для идентификации и количественного определения L-карнитина. Полученное нами содержание L-Кр в напитке XXI Power “L-КАРНИТИН” методом мицеллярной ТСХ без концентрирования (неподвижная фаза: Сорбфил; ПФ: ЦПХ (1.0 × 10–3 М)–вода) составило (2.3 ± 0.2) мг/мл (n = = 3, P = 0.95, sr = 0.10). Видно, что найденное содержание практически соответствует заявленному, что свидетельствует о правильности определения L-карнитина методом мицеллярной ТСХ.
* * *
Таким образом, предложены новые подвижные фазы (водно-органическая на основе бутанола-2 и мицеллярная на основе 0.001 М раствора хлорида цетилпиридиния), новая неподвижная фаза на основе Силуфола для тонкослойной хроматографии L-карнитина и выявлены преимущества мицеллярной ПФ. Показано, что для концентрирования L-карнитина может быть использован метод магнитной твердофазной экстракции, основанный на сорбции аналита магнитными наночастицами магнетита, модифицированными бромидом цетилтриметиламмония. При варьировании рН раствора, массы сорбента, времени сорбции, природы и объема элюента найдены оптимальные условия количественной сорбции и десорбции L-карнитина. Разработана методика денситометрического определения L-карнитина с помощью мицеллярной ТСХ в напитке для спорта XXI Power “L-КАРНИТИН”.
Работа выполнена при поддержке РНФ, проект № 21-13-00267.
Список литературы
Bremer J. Carnitine — Metabolism and functions // Physiol. Rev. 1983. V. 63. P. 1420. https://doi.org/10.1152/physrev.1983.63.4.1420
Pekala J., Patcowska-Sokola B., Bodkowski R., Jamroz D., Nowakowski P., Lochynski S., Librowski T. L-Carnitine – Metabolic functions and meaning in humans life // Curr. Drug. Metabol. 2011. V. 12. P. 667. https://doi.org/10.2174/138920011796504536
Adeva-Andany M.M., Calvo-Castro I., Carlos Fernandez-Fernandez C., Donapetry-Garcıa C., Pedre-Pineiro A.M. Significance of L-carnitine for human health // IUBMB Life. 2017. V. 69. № 8. P. 578. https://doi.org/10.1002/iub.1646
Fathizadeh H., Milajerdi A., Reiner Ž., Amirani E., Asemi Z., Mansournia M.A., Hallajzadeh J. The effects of L-carnitine supplementation on indicators of inflammation and oxidative stress: A systematic review and meta-analysis of randomized controlled trials // J. Diabetes Metab. Disord. 2020. V. 19. P. 1879. https://doi.org/10.1007/s40200-020-00627-9
Сизова Ж.М., Ших Е.В., Махова А.А. Применение L-карнитина в общей врачебной практике // Терапевтич. архив. 2019. Т. 91. № 1. С. 114. (Sizova Z.M., Shikh E.V., Makhova A.A. Significance of L-carnitine in internal medicine // Therapeut. Arch. 2019. V. 91. № 1. P. 114.)
Ефимова Е.В., Гуськова Т.А., Копелевич В.М., Гунар В.И. Ацетил-L-карнитин: биологические свойства и клиническое применение (обзор) // Хим.-фарм. журн. 2002. Т. 36. № 3. С. 3. (Efimova E.V., Gus’kova T.A., Kopelevich V.M., Gunar V.I. Acetyl-L-carnitine: Biological properties and clinical application (A review) // Pharm. Chem. J. 2002. V. 36. № 3. P. 111.)
Sawicka A.K., Hartmane D., Lipinska P., Wojtowicz E., Lysiak-Szydlowska W., Olek R.A. L-Carnitine supplementation in older women. A pilot study on aging skeletal muscle mass and function // Nutrients. 2018. V. 10. № 2. P. 255. https://doi.org/10.3390/nu10020255
Sawicka A.K., Renzi G., Olek R.A. The bright and the dark sides of L-carnitine supplementation: A systematic review // J. Int. Soc. Sports Nutr. 2020. V. 17. P. 49. https://doi.org/10.1186/s12970-020-00377-2
Насирова У.Ф., Тастанова Р.М., Пак А.А., Шарипова М.К. Ранняя диагностика карнитиновой недостаточности у недоношенных новорожденных // Рос. вестн. перинатол. и педиатрии 2018. Т. 63. № 3. С. 39. (Nasirova U.F., Tastanova R.M., Pak A.A., Sharipova M.K. Early diagnosis of carnitine insufficiency in premature infants // Russ. Bull. Perinat. Pediatr. 2018. V. 63. № 3. P. 39.) https://doi.org/10.21508/1027-4065-2018-63-3-39-44
Seline K.G., Johein H. The determination of L-carnitine in several food samples // Food Chem. 2007. V. 105. № 2. P. 793. https://doi.org/10.1016/j.foodchem.2007.01.058
Marzo A., Cardace G., Monti N., Muck S., Arrigoni Martelli E. Chromatographic and non-chromatographic assay of L-carnitine family components // J. Chromatogr. 1990. V. 527. P. 247. https://doi.org/10.1016/S0378-4347(00)82109-X
Deufel T. Determination of L-carnitine in biological fluids and tissues // J. Clin. Chem. Clin. Biochem. 1990. V. 28. № 5. P. 307.
Marzo A., Curti S. L-carnitine moiety assay: An up-to-date reappraisal covering the commonest methods for various applications // J. Chromatogr. B. 1997. V. 702. P. 1. https://doi.org/10.1016/S0378-4347(97)00376-9
Dabrowska M., Starek M. Analytical approaches to determination of carnitine in biological materials, foods and dietary supplements // Food Chem. 2014. V. 142. P. 220. https://doi.org/10.1016/j.foodchem.2013.06.137
Mansour F.R., Wei W., Danielson N.D. Separation of carnitine and acylcarnitines in biological samples: A review // Biomed. Chromatogr. 2013. V. 27. 10. P. 1339. https://doi.org/10.1002/bmc.2995
Minkler P.E., Ingalls S.T., Hoppel C.L. Strategy for the isolation, derivatization, chromatographic separation, and detection of carnitine and acylcarnitines // Anal. Chem. 2005. V. 77. № 5. P. 1448. https://doi.org/10.1021/ac0487810
Li K., Sun Q. Simultaneous determination of free and total carnitine in human serum by HPLC with UV detection // J. Chromatogr Sci. 2010. V. 48. P. 371. https://doi.org/10.1093/chromsci/48.5.371
Park J.M., Koh J.H., Kim J.M. Determination of L-carnitine in infant powdered milk samples after derivatization // Food Sci. Anim. Resour. 2021. V. 41. № 4. P. 731. https://doi.org/10.5851/kosfa.2021.e23
Захарычев В.В., Испирян А.З., Смелкова О.И., Сысуева С.В. Методика раздельного количественного определения содержания L- и D-изомеров карнитина // Ветеринария. 2021. № 8. С. 59. (Zakharychev V.V., Ispiryan A.Z., Smelkova O.I., Sysueva S.V. Development of a method for differential quan-titative determination of L-carnitine and D-carnitine // Veterinaria. 2021. № 8. P. 59.) https://doi.org/10.30896/0042-4846.2021.24.8.59-64
Kakou A., Megoulas N.C., Koupparis M.A. Determination of L-carnitine in food supplement formulations using ion-pair chromatography with indirect conductometric detection // J. Chromatogr. A. 2005. V. 1069. P. 209. https://doi.org/10.1016/j.chroma.2005.02.021
Nie L.-J., Liang J., Shan F., Xu Y.-Y., Yan C.-Y., Zhou X., Cheng Z.-Y., Xia Q.-R. A UPLC–MS/MS method for determination of endogenous l-carnitine and acetyl-l-carnitine in serum of patients with depression // Biomed. Chrom. 2021. V. 35. № 3. Article e4991. https://doi.org/10.1002/bmc.4991
Kong Y., Yang G., Chen S., Hou Z., Du X., Li H., Kong L. Rapid and sensitive determination of L-carnitine and acetyl-L-carnitine in liquid milk samples with capillary zone electrophoresis using indirect UV detection // Food Anal. Methods. 2018. V. 11. № 1. P. 170. https://doi.org/10.1007/s12161-017-0987-y
Wei D., Liu J., Guo M., Zhu Y. Determination of betaine, L-carnitine and choline in human urine using a self-packed column and column-switching ion chromatography with non-suppressed conductivity detection // J. Separ. Sci. 2017. V. 40. № 21. P. 4246. https://doi.org/10.1002/jssc.201700545
Manjón A., Obón J.M., Iborra J.L. Determination of L-carnitine by flow injection analysis with NADH fluorescence detection // Anal. Biochem. 2000. V. 281. № 2. P. 176. https://doi.org/10.1006/abio.2000.4573
Hardy D.T., Preece M.A., Green A. Determination of plasma free carnitine by electrospray tandem mass spectrometry // Annals Clin. Biochem. 2001. V. 38. P. 665. https://doi.org/10.1258/0004563011901073
Schäfer J., Reichmann H. A spectrophotometric method for the determination of free and esterified carnitine // Clin. Chim. Acta. 1989. V. 182. № 1. P. 87. https://doi.org/10.1016/0009-8981(89)90152-6
Serdar M.A., Kurt Ü., Bayraktar Ü., Kenar L., Zelük F., Onur Ü., Kutluay T. Optimization of fluorometric measurement of free and total carnitine in serum // Turk. J. Med. Sci. 2001. V. 31. P. 309.
Isaguirre A.C., Olsina R.A., Martinez L.D., Lapierre A.V., Cerutti S. Development of solid phase extraction strategies to minimize the effect of human urine matrix effect on the response of carnitine by UPLC-MS/MS // Microchem. J. 2016. V. 129. P. 362. https://doi.org/10.1016/j.microc.2016.07.018
Chen Y.-C., Tsai C.-J., Feng C.-H. Fluorescent derivatization combined with aqueous solvent-based dispersive liquid-liquid microextraction for determination of butyrobetaine, L-carnitine and acetyl-L-carnitine in human plasma // J. Chromatogr. A. 2016. V. 1464. P. 32. https://doi.org/10.1016/j.chroma.2016.08.030
Егунова О.Р., Константинова Т.А., Штыков С.Н. Магнитные наночастицы магнетита в разделении и концентрировании // Изв. Сарат. ун-та. Новая серия. Серия: Химия. Биология. Экология. 2014. Т. 14. №. 4. С. 27. (Egunova O.R., Konstantinova T.A.,. Shtykov S.N. Magnetic Nanoparticles in Separation and Preconcentration // Izv. Saratov Univ. (N.S.). Ser. Chemistry. Biology. Ecology. 2014. V. 14. P. 27.) https://doi.org/10.18500/1816-9775-2014-14-4-27-35
Толмачева В.В., Апяри В.В., Кочук Е.В., Дмитриенко С.Г. Магнитные сорбенты на основе наночастиц оксидов железа для выделения и концентрирования органических соединений // Журн. аналит. химии. 2016. Т. 71. № 4. С. 339. (Tolmacheva V.V., Apyari V.V., Kochuk E.V., Dmitrienko S.G. Magnetic adsorbents based on iron oxide nanoparticles for the extraction and preconcentration of organic compounds // J. Anal. Chem. 2016. V. 71. № 4. P. 321.) https://doi.org/10.1134/S1061934816040079
Егунова О.Р., Решетникова И.С., Казимирова К.О., Штыков С.Н. Магнитная твердофазная экстракция и флуориметрическое определение некоторых фторхинолонов // Журн. аналит. химии. 2020. Т. 75. № 1. С. 31. (Egunova O.R., Reshetnikova I.S., Kazimirova K.O., Shtykov S.N. Magnetic solid-phase extraction and fluorimetric determination of some fluoroquinolones // J. Anal. Chem. 2020. V. 75. № 1. P. 24.) https://doi.org/10.1134/S1061934820010062
Пряжников Д.В., Кубракова И.В. Магнитные наноразмерные материалы с модифицированной поверхностью: получение и исследование структуры, состава и свойств // Журн. аналит. химии. 2021. Т. 76. № 6. С. 496. (Pryazhnikov D.V., Kubrakova I.V. Surface-modified magnetic nanoscale materials: Preparation and study of their structure, composition, and properties // J. Anal. Chem. 2021. V. 76. № 6. P. 685.) https://doi.org/10.1134/S1061934821060095
Моходоева О.Б., Максимова В.В., Дженлода Р.Х., Шкинев В.М. Модифицированные ионными жидкостями магнитные наночастицы в анализе объектов окружающей среды // Журн. аналит. химии. 2021. Т. 76. № 6. С. 483. (Mokhodoeva O.B., Maksimova V.V., Dzhenloda R.Kh., Shkinev V.M. Magnetic nanoparticles modified by ionic liquids in environmental analysis // J. Anal. Chem. 2021. V. 76. 6. P. 675.) https://doi.org/10.1134/S1061934821060058
Dąbrowska M., Sieczka E., Starek M. TLC assay of L-carnitine in dietary supplements // J. Planar Chromatogr. 2012. V. 25. № 5. P. 450. https://doi.org/10.1556/jpc.25.2012.5.11
Сумина Е.Г., Штыков С.Н., Тюрина Н.В. Поверхностно-активные вещества в тонкослойной хроматографии // Журн. аналит. химии. 2003. Т. 58. № 8. С. 809. (Sumina E.G., Shtykov S.N., Tyurina N.V. Surfactants in thin layer chromatography // J. Anal. Chem. 2003. V. 58. № 8. P. 720.) https://doi.org/10.1023/A:1025027409149
Mohammad A., Haq N. TLC separation of amino acids with a green mobile phase // J. Planar Chromatogr. 2010. V. 23. № 4. P. 260. https://doi.org/10.1556/JPC.23.2010.4.4
Bhawani S.A., Albishri H.M., Khan Z.A., Ibrahim M.N.M, Mohammad A. Surfactant modified/mediated thin-layer chromatographic systems for the analysis of amino acids // J. Anal. Methods Chem. 2013. V. 2013. 973280. https://doi.org/10.1155/2013/973280
Штыков С.Н., Сумина Е.Г., Угланова В.З., Сорокина О.Н. Тонкослойная хроматография некоторых аминокислот на силикагеле в водно-органических и модифицированных мицеллярных подвижных фазах // Журн. аналит. химии. 2017. Т. 72. № 8. С. 742. (Shtykov S.N., Sumina E.G., Uglanova V.Z., Sorokina O.N. Thin-layer chromatography of some amino acids on silica in aqueous–organic and modified micellar mobile phases // J. Anal. Chem. 2017. V. 72. № 8. P. 870.) https://doi.org/10.1134/S1061934817080111
Shtykov S.N., Sumina E.G., Smushkina E.V., Tyurina N.V. Dynamic and static modification of the stationary phases with surfactants in TLC: A comparative study // J. Planar Chromatogr. – Modern TLC. 2000. V. 13. № 3. P. 182.
Казимирова К.О., Хабибуллин В.Р., Решетникова И.С., Егунова О.Р., Штыков С.Н. Концентрирование пищевых азокрасителей Е110 и Е124 на наночастицах магнетита, модифицированных ЦТАБ // Изв. Сарат. ун-та. Нов. сер. Сер. Химия. Биология. Экология. 2017. Т.17. № 2. С. 138. (Kazimirova K.O., Khabibullin V.R., Reshetnikova I.S., Egunova O.R., Shtykov S.N. Preconcentration of Е110 and Е124 food azodyes on magnetite nanoparticles modified by CTAB // Izv. Saratov Univ. (N.S.). Ser. Chemistry. Biology. Ecology. 2017. V. 17. №. 2. P. 138.) https://doi.org/10.18500/1816-9775-2017-17-2-138-142
Штыков С.Н., Сумина Е.Г., Тюрина Н.В. Расчет коэффициентов межфазного распределения органических реагентов в мицеллярной ТСХ // Журн. аналит. химии. 2002. Т. 57. № 4. С. 383. (Shtykov S.N., Sumina E.G., Tyurina N.V. Calculation of partition coefficients of organic reagents in micellar thin-layer chromatography // J. Anal. Chem. 2002. V. 57. № 4. P. 322.) https://doi.org/10.1023/A:1014950314681
Ворожейкин С.Б., Башко Е.С., Штыков С.Н. Тонкослойная хроматография аминокислот в мицеллярных подвижных фазах на силикагеле // Сорбционные и хроматогр. процессы. 2011. Т. 11. № 6. С. 840.
Сорокоумов П.Н., Савельева Е.И., Каракашев Г.В., Копейкин В.А., Радилов А.С. Определение мельдония, гамма-бутиробетаина и карнитина в плазме крови методом высокоэффективной жидкостной хроматографии c масс-селективным детектированием // Разработка и регистрация лекарственных средств. 2016. № 1. С. 176. (Sorokoumov P.N., Savelieva E.I., Karakashev G.V., Kopeikin V.A., Radilov A.S. Determination of meldonium, γ-butyrobetaine, and carnitine in blood plasma by high-performance liquid chromatography with mass-selective detection // Drug development and registration. 2016. № 1. P. 176.)
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии