

Журнал общей биологии, 2023, T. 84, № 2, стр. 83-97
Роль человеческого сывороточного альбумина в профилактике и лечении болезни Альцгеймера
М. П. Шевелёва 1, Е. И. Дерюшева 1, Е. Л. Немашкалова 1, А. В. Мачулин 2, Е. А. Литус 1, *
1 Институт биологического приборостроения с опытным производством РАН, обособленное подразделение ФИЦ “Пущинский научный центр биологических исследований РАН”
142290 Московская обл., Пущино, Пр-т Науки, 3, Россия
2 Институт биохимии и физиологии микроорганизмов им. Г.К. Скрябина РАН, обособленное подразделение ФИЦ “Пущинский научный центр биологических исследований РАН”
142290 Московская обл., Пущино, Пр-т Науки, 3, Россия
* E-mail: ealitus@gmail.com
Поступила в редакцию 19.08.2022
После доработки 08.01.2023
Принята к публикации 22.03.2023
- EDN: RAPODG
- DOI: 10.31857/S0044459623020069
Аннотация
Болезнь Альцгеймера (БА) была и остается основной причиной развития деменции у возрастных пациентов. Данное нейродегенеративное заболевание характеризуется прогрессивным течением и относится к группе социально значимых. Существует несколько гипотез развития БА: тау-гипотеза, амилоидная гипотеза, холинергическая гипотеза, гипотезы окислительного стресса и воспаления. Отсутствие общепринятого представления об этиологии и патогенезе БА препятствует разработке новых эффективных способов ее лечения и профилактики. В клинической практике широко используются ингибиторы холинэстеразы, облегчающие симптомы заболевания, но не влияющие на его течение. В 2021 г. впервые был одобрен препарат для проведения патогенетической терапии БА (адуканумаб), способствующий снижению содержания β-амилоидного пептида (Аβ) в головном мозге пациентов. Другим перспективным подходом к терапии БА, направленным на выведение Аβ из центральной нервной системы пациента, является воздействие на человеческий сывороточный альбумин (ЧСА), который переносит 90% Аβ в сыворотке крови и 40–90% Аβ в цереброспинальной жидкости. В клинической практике уже был апробирован и показал свою эффективность плазмаферез с заменой собственного ЧСА на очищенный терапевтический препарат альбумина. Еще одним вариантом такого подхода является усиление взаимодействия ЧСА с Аβ посредством воздействия экзогенных и эндогенных лигандов ЧСА, таких как серотонин, ибупрофен и некоторые ненасыщенные жирные кислоты. Исследования in vivo подтверждают ассоциацию данной группы лигандов с патогенезом БА. Перечисленные вещества относятся к хорошо изученным естественным метаболитам или лекарственным препаратам, что существенно упрощает разработку новых методов терапии и профилактики БА с их использованием. В целом, новое направление научных исследований, посвященных изучению ЧСА в качестве переносчика и депо Аβ в крови и цереброспинальной жидкости, позволит расширить наши представления о метаболизме Аβ и его роли в патогенезе БА.
БОЛЕЗНЬ АЛЬЦГЕЙМЕРА: ОСНОВНЫЕ ТЕОРИИ ПАТОГЕНЕЗА И ПОДХОДЫ К ТЕРАПИИ
Деменция – патологическое состояние, характеризующееся нарушением мыслительной и стойким снижением познавательной деятельности. При этом пациент утрачивает ранее приобретенные знания и навыки, а также частично или полностью лишается способности к обучению (Arvanitakis et al., 2019). Деменцию относят к основным причинам инвалидизации и социальной дезадаптации пожилых людей: по данным ВОЗ, доля пациентов с деменцией среди населения в возрасте 60 лет и старше составляет 6–8%. Цифры заболеваемости неуклонно растут: согласно прогнозам, численность пациентов с деменцией к 2050 г. увеличится более чем втрое и составит 152 млн человек (GBD 2019..., 2022). В 60–70% случаев причиной развития деменции является болезнь Альцгеймера (БА) (по данным ВОЗ), относящаяся к группе нейродегенеративных заболеваний и характеризующаяся прогрессивным течением (Kumar et al., 2022). Несмотря на множество исследований, посвященных изучению этой патологии, до сих пор нет полного представления об ее этиологии и патогенезе и не разработано эффективной общепринятой схемы лечения пациентов с диагнозом БА. В то же время данная категория пациентов требует особого ухода и поддерживающей терапии в течение длительного периода, что обуславливает большую социальную и экономическую значимость БА (постановлением Правительства РФ № 715 от 01.12.04 отнесено к категории социально значимых заболеваний).
Существует несколько теорий развития БА: тау-гипотеза, амилоидная гипотеза, холинергическая гипотеза, гипотезы окислительного стресса и воспаления, – каждая рассматривает соответствующее звено патогенеза в качестве решающего фактора в течение заболевания.
Основными патоморфологическими признаками БА, локализованными в головном мозге пациентов, считаются отложения β-амилоидного пептида (Аβ) в виде синильных бляшек, нейрофибриллярные клубки и массовая гибель нейронов (Cheignon et al., 2018; Sheppard, Coleman, 2020). Аβ образуется из трансмембранного белка – предшественника бета-амилоида. Основными его разновидностями являются пептиды длинной 40 и 42 аминокислоты (Аβ40 и Аβ42) (Murphy, LeVine, 2010). Амилоидная гипотеза развития БА предполагает, что именно накопление Аβ в головном мозге пациента приводит к образованию нейрофибриллярных клубков (Sadigh-Eteghad et al., 2015), воспалению (Meraz-Ríos et al., 2013), нарушению синаптической передачи (Shankar, Walsh, 2009) и гибели нейронов (Moreira et al., 2010), что сопровождается характерной клинической картиной. О первостепенной роли Аβ в течение БА говорит и то, что амилоидные отложения появляются до развития клинической картины заболевания (Vandesquille et al., 2014), а нейротоксичность различных форм Аβ подтверждена in vivo и in vitro (Carrillo-Mora et al., 2014). Наследственные формы БА связывают с нарушением метаболизма Аβ и в первую очередь с ускорением его продукции (Cheignon et al., 2018). В то же время было показано, что у пациентов с диагнозом БА нарушено выведение Аβ из центральной нервной системы (ЦНС), снижены концентрация и активность ферментов, отвечающих за протеолиз Аβ (Wang et al., 2006; Zhang et al., 2018). Предполагается, что нарушение выведения Аβ из ЦНС приводит к его накоплению и развитию спорадических форм БА, составляющих более 90% всех случаев (Bali et al., 2012). Одним из основных доводов против амилоидной гипотезы до недавнего времени было отсутствие лекарственных средств с подтвержденной эффективностью для лечения БА, направленных против Аβ, его мономерных, олигомерных и фибриллярных форм. Данный аргумент был частично снят в 2021 г. после регистрации Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов (Food and Drug Administration – FDA, США) антитела к Аβ, препарата адуканумаб, применение которого приводило к снижению отложений Аβ в ЦНС пациентов с диагнозом БА и клиническим улучшениям (Sevigny et al., 2016; Tampi et al., 2021). В то же время FDA потребовало проведения пострегистрационной фазы испытаний препарата для формирования окончательного решения о его клинической значимости (https://www.fda.gov/drugs/news-events-human-drugs/fdas-decision-approve-new-treatment-alzheimers-disease).
Еще один признак БА − внутриклеточные нейрофибриллярные клубки (Cheignon et al., 2018; Sheppard, Coleman, 2020) – представляет собой внутриклеточные скопления гиперфосфорилированного тау-белка, который в норме отвечает за поддержание цитоскелета нейронов (Metaxas, Kempf, 2016). Тау-белок подвергается множеству модификаций, включая фосфорилирование, метилирование, ацетилирование и др. (Du et al., 2018). В патологических условиях процесс гиперфосфорилирования тау-белка приводит к нарушению его ассоциации с микротрубочками, что влечет за собой их дестабилизацию и нарушение аксонального транспорта (Alonso et al., 1994; Rodríguez-Martín et al., 2013), а в дальнейшем к разрушению синапсов и нарушению взаимодействия между нейронами (Spires-Jones, Hyman, 2014). Данные о наличии корреляции между накоплением нейрофибриллярных клубков и степенью тяжести деменции (Brier et al., 2016) сделали тау-белок важной мишенью для разработки подходов к лечению БА. Среди них можно выделить разработку вакцин против гиперфосфорилированного тау-белка, блокаторы его агрегации, стабилизаторы микротрубочек (Du et al., 2018). Многие из этих разработок не показали себя в клинических испытаниях, и ни одна из них не была одобрена для внедрения в практику (Du et al., 2018; Mullard, 2021). Более того, у некоторых пациентов с диагнозом БА не удается обнаружить нейрофибриллярные клубки (Tiraboschi et al., 2004), и к настоящему времени не выявлены генетические формы БА, обусловленные мутациями в гене тау-белка (Goedert, Spillantini, 2001; Poorkaj et al., 2001). Все формы деменции, вызванные мутациями в гене тау-белка, классифицируют как фронтотемпоральную деменцию.
Перечисленные характерные патоморфологические признаки БА в ЦНС пациентов сопровождаются активацией микроглии и воспалением, которые играют важную роль в развитии БА. В настоящий момент показано, что персистирующий воспалительный процесс приводит к усугублению нейродегенеративных изменений, поддерживает и усиливает накопление нейрофибриллярных клубков и амилоидных бляшек (Kinney et al., 2018). Большое количество как про-, так и противовоспалительных цитокинов и медиаторов ассоциированы с патогенезом БА (Azizi et al., 2015; Kinney et al., 2018). С другой стороны, несмотря на данные эпидемиологических исследований о снижении риска развития БА при длительном приеме нестероидных противовоспалительных препаратов (Vlad et al., 2008; Wang et al., 2015; Rivers-Auty et al., 2020) и положительные результаты, полученные для этой группы лекарственных средств на животных моделях (Lim et al., 2000; Yan et al., 2003; McKee et al., 2008), данные клинических исследований терапевтических эффектов нестероидных противовоспалительных препаратов при лечении БА неоднозначны (Hayden et al., 2007; Tschanz et al., 2013; Miguel-Álvarez et al., 2015; Ali et al., 2019). Возникшие противоречия можно объяснить неправильным выбором популяции для проведения клинических исследований, небольшой численностью групп и недостаточным периодом наблюдения (Ali et al., 2019). Было показано, что для положительного эффекта нестероидных противовоспалительных средств имеет значение возраст пациента, наличие у него APOE ε4 аллели и длительность приема препарата (Hayden et al., 2007; Tschanz et al., 2013; Ali et al., 2019). Некоторыми исследователями высказывается предположение, что положительного эффекта от приема нестероидных противовоспалительных средств можно добиться только на ранних стадиях БА, в период начала накопления Аβ в головном мозге пациентов (Ali et al., 2019). В противном случае лечение такими препаратами может нанести вред пациенту из-за ингибирующих эффектов на активированную микроглию, которая на ранних этапах активации препятствует отложению Аβ в головном мозге (Kinney et al., 2018; Ali et al., 2019). Для окончательных выводов о клинической значимости использования нестероидных противовоспалительных средств для терапии и профилактики БА необходимо проведение длительных клинических исследований с большим числом участников. Кроме того, необходимо оценить вклад различных факторов, таких как генотип и возраст пациентов, в наблюдаемые эффекты (Ali et al., 2019).
Еще одним важным патогенетическим механизмом БА является окислительный стресс. Данный процесс не только связан с перекисным окислением липидов и окислением белков, но и задействован в реализации токсических эффектов Аβ (Butterfield, Lauderback, 2002; Gibson et al., 2004; Butterfield et al., 2007; Cheignon et al., 2018; Du et al., 2018). В то же время окислительный стресс − неспецифический патофизиологический механизм, который участвует в развитии не только нейродегенеративных, но и кардиоваскулярных и раковых заболеваний (Pizzino et al., 2017). И, несмотря на его явную связь с прогрессией заболевания (Christen, 2000), окислительный стресс не является ведущим фактором патогенеза БА. По этой причине антиоксиданты изучают на предмет возможного использования в терапии БА (Christen, 2000; Gella, Durany, 2009), но скорее как часть комбинированной терапии, а не в качестве самостоятельной стратегии лечения.
Одним из первых общепринятых подходов к лечению БА было использование ингибиторов холинэстеразы (Summers et al., 1981, 1986; Brinkman, Gershon, 1983). Выбор данной стратегии в лечении БА был обусловлен участием холинергических нейронов в таких важных физиологических процессах, как память, обучение, внимание, а также наличием корреляции между повреждением холинергических нейронов и степенью когнитивных нарушений при БА (Du et al., 2018). В то же время использование ингибиторов холинэстеразы приносит временный эффект и неспособно остановить развитие заболевания (Sharma, 2019).
Несмотря на многолетние споры и попытки сформулировать альтернативную теорию патогенеза БА, амилоидная гипотеза все еще остается актуальной. Со временем она дополнялась и перерабатывалась, что помогло найти новые подходы к лечению БА. Все больше исследований последних лет направлены на изучение механизмов выведения Aβ из ЦНС (Deane et al., 2009; Zhang et al., 2013; McCormick et al., 2021), в том числе за счет периферических транспортных белков, основным из которых является человеческий сывороточный альбумин (ЧСА) (Biere et al., 1996; Kuo et al., 2000; Choi et al., 2017).
ЧЕЛОВЕЧЕСКИЙ СЫВОРОТОЧНЫЙ АЛЬБУМИН: ДЕПОНИРОВАНИЕ И ТРАНСПОРТ Aβ
Человеческий сывороточный альбумин (ЧСА) является наиболее распространенным белком плазмы (его концентрация около 0.6 мМ), при этом более чем 90% Aβ связывается с ЧСА в сыворотке крови и около 40–90% – в цереброспинальной жидкости (Milojevic, Melacini, 2011; Stanyon, Viles, 2012; Algamal et al., 2013), хотя концентрация ЧСА в ней существенно ниже, чем в сыворотке: около 3 мкM (Bohrmann et al., 1999; Schilde et al., 2018).
В литературе есть подтверждения, что ЧСА связывает мономеры Aβ со стехиометрией 1 : 1 и константой диссоциации, по разным данным, от 10–9–10–8 (Litus et al., 2019, 2021) до 10–6–10–5 М (Kuo et al., 2000; Rózga et al., 2007; Stanyon, Viles, 2012). Такое различие значений констант обусловлено, по-видимому, как используемыми методами определения аффинности, так и в большой степени “предысторией” образца Aβ (Suvorina et al., 2015). Различные способы предварительной солюбилизации Aβ для получения в растворе его мономерной формы – обработка растворами сильных кислот и щелочей (Milojevic et al., 2009; Litus et al., 2019, 2021, 2022) либо апротонными растворителями, 1,1,1,3,3,3-гексафтор-2-пропанолом и диметилсульфоксидом (Costa et al., 2012; Wang et al., 2016) – изменяют константу диссоциации комплекса ЧСА–мономер Aβ от 10–9 M до отсутствия детектируемого взаимодействия. Пробоподготовка, очевидно, оказывает влияние на распределение конформационных изомеров в образце – статистический клубок, β-складки либо α-спираль, что в итоге влияет не только на константу диссоциации комплекса ЧСА–мономер Aβ, но также на морфологию образующихся олигомеров (Kirkitadze et al., 2001; Suvorina et al., 2015). ЧСА также эффективно взаимодействует с олигомерами Aβ с константой диссоциации порядка 1–100 нМ (Milojevic et al., 2007, 2009; Milojevic, Melacini, 2011), протофибриллами (Milojevic et al., 2009; Algamal et al., 2013, 2017) и фибриллами (Xie, Guo, 2020).
Экспериментальные и молекулярно-динамические исследования (Choi et al., 2017) показали, что бороздка ЧСА между доменами 1 и 3 является наиболее вероятным местом связывания мономера Aβ (рис. 1 ). Процесс связывания сопровождается преобразованием структуры Aβ из клубка в α-спираль; при этом заметных структурных изменений в ЧСА не наблюдалось (Choi et al., 2017). Нами был предсказан потенциальный вторичный сайт связывания ЧСА с мономерной формой Aβ (Litus et al., 2022). ЧСА в первую очередь взаимодействует с С-концевыми остатками мономера Aβ (сегмент с 31 по 40–42 аминокислотный остаток, а. о.) с дополнительными взаимодействиями по центральному сегменту пептида (с 12 по 24 а. о.) (Algamal et al., 2013; Litus et al., 2022).
Рис. 1.
Структура ЧСА и Aβ: а – структура ЧСА (PDB код: 1AO6), б – структура β-амилоидного пептида 1-40 (PDB код: 2LFM), в – структура β-амилоидного пептида 1-42 (PDB код: 1IYT).
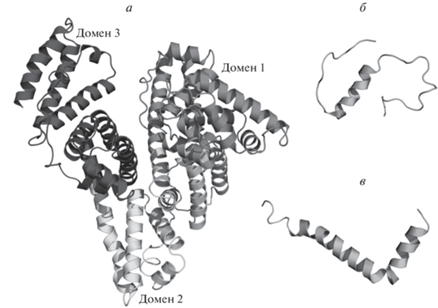
Домены 1 и 3 также содержат потенциальные сайты связывания олигомеров Aβ (Zhao, Guo, 2021). В частности, регион 494–515 а. о. во втором субдомене домена 3 идентифицирован как предполагаемый сайт связывания олигомера Aβ (Algamal et al., 2013).
Интересно, что ЧСА не только связывает и изолирует большую часть Aβ в плазме, но также способствует транспортировке Aβ из спинномозговой жидкости в плазму крови (Kuo et al., 2000; Boada et al., 2009; Stanyon, Viles, 2012; Ezra et al., 2016). По литературным данным, прямой транспорт Aβ через гематоэнцефалический барьер составляет 25% его клиренса у когнитивно нормальных людей (Roberts et al., 2014). При этом исследования, проведенные на животных моделях, показали, что период полувыведения Aβ между цереброспинальной жидкостью и плазмой составляет около 30 мин (Ghersi-Egea et al., 1996; Poduslo et al., 1999; Shibata et al., 2000). Эти результаты свидетельствуют о том, что существует равновесие между Aβ в плазме и в цереброспинальной жидкости. При БА создается новое равновесие, при котором Aβ не только попадает в плазму, но и откладывается в ЦНС (DeMattos et al., 2002). Такое динамическое равновесие можно изменить с помощью терапевтических вмешательств, направленных на усиление клиренса Aβ из ЦНС и его депонирование в сыворотке крови посредством образования комплекса с ЧСА (Matsuoka et al., 2003; Boada et al., 2017).
Перечисленные факты указывают на первостепенное значение ЧСА в метаболизме Aβ. По этой причине ЧСА рассматривается как терапевтическая мишень для лечения БА (Boada et al., 2017; Cuberas-Borrós et al., 2018; Menendez-Gonzalez, Gasparovic, 2019).
ВЛИЯНИЕ ЧЕЛОВЕЧЕСКОГО СЫВОРОТОЧНОГО АЛЬБУМИНА НА ПРОЦЕСС ОБРАЗОВАНИЯ ФИБРИЛЛ Aβ
Для понимания путей, по которым должна идти разработка подходов к терапии, основанных на взаимодействии ЧСА с Aβ, нужно изучить механизмы влияния альбумина на образование токсичных форм Aβ. Это необходимо как для целенаправленного воздействия на ингибирующую способность ЧСА, так и для прогнозирования последствий его возможных биохимических модификаций, возникающих в результате мутаций, сопутствующих заболеваний и др.
Механизм образования амилоидных фибрилл как in vitro (Kirkitadze et al., 2001; Bitan et al., 2003), так и in vivo (Pitschke et al., 1998; Gong et al., 2003; Kayed et al., 2003) разработан достаточно подробно. Фибриллы формируются последовательностью параллельных либо антипараллельных β-листов (Fändrich, 2007; Qiang et al., 2012). Начальной стадией процесса является конформационная перестройка мономерного пептида, сопровождающаяся переходом преимущественно неупорядоченной структуры (Zhang et al., 2000; Baumketner et al., 2006) в β-складку (Kirschner et al., 1986). Такие мономеры более склонны к олигомеризации и формируют мультимерные и олигомерные структуры с различным количеством вовлеченных мономеров (от димеров до сферических олигомерных структур диаметром до десятков нанометров (Bitan et al., 2003; Bernstein et al., 2005)), все еще растворимые (Kayed et al., 2003), но являющиеся зародышами последующего процесса формирования фибрилл. Следующей стадией является образование протофибрилл, протекающее как по механизму соединения нескольких олигомеров, так и удлинения путем присоединения мономеров к C-концу растущей цепи, и далее образование зрелых фибрилл длиной до нескольких микрон и толщиной 5–15 нм (Fändrich et al., 2009). Переход протофибрилл в зрелые фибриллы сопровождается не только увеличением размеров, но и существенной структурной перестройкой, ростом упорядоченности и гидрофобности вследствие увеличения доли фибриллярных β-структур (Wang et al., 2016), прочно связанных внутримолекулярными неполярными и водородными связями. Это в итоге обусловливает крайне низкую растворимость зрелых фибрилл (Kirschner et al., 1986).
Механизмы влияния ингибирующего агента в зависимости от стадии фибриллообразования можно разделить на два типа (Milojevic et al., 2009): 1) непосредственное взаимодействие ингибитора с мономером, приводящее к снижению свободной концентрации последнего и стабилизации его мономерного состояния, препятствующим зародышеобразованию или присоединению мономера к растущей цепи – модель “стабилизации мономера”; 2) взаимодействие с уже образовавшимися протофибриллами, препятствующее их удлинению и переходу в зрелые фибриллы – модель “мономер-конкурент”.
Как указывалось выше, ЧСА является основным депо Aβ как в плазме крови, так и в цереброспинальной жидкости, регулируя концентрацию свободного Aβ. Исследования подтверждают факт ингибирующего действия ЧСА на кинетику фибриллообразования (Bohrmann et al., 1999; Reyes Barcelo et al., 2009). Снижение концентрации ЧСА, наблюдаемое с возрастом и при воспалительных процессах, приводит к увеличению концентрации Aβ и коррелирует с увеличением риска БА (Llewellyn et al., 2010).
Этапом, предшествующим зародышеобразованию, является формирование в Aβ склонных к агрегации β-шпилек, нарушаемое в присутствии ЧСА. Более того, ЧСА препятствует межмолекулярному взаимодействию, необходимому для образования этих β-шпилек (Xie, Guo, 2020). Aβ, связавшись своим гидрофобным ядром с поверхностью ЧСА и будучи сам отрицательно заряженным, одновременно испытывает электростатическое отталкивание от одноименно заряженных областей альбумина (Algamal et al., 2013). Равновесие сил притяжения и отталкивания стабилизирует более протяженную структуру Aβ в отличие от β-складчатой, причем увеличение как положительного, так и отрицательного поверхностного заряда ЧСА путем химической модификации усиливает его ингибирующий эффект (Rayner, Hasking, 1986; Xie et al., 2014), поскольку, в отличие от немодифицированного ЧСА, селективно связывающего мономер посредством преимущественно гидрофобных взаимодействий, ЧСА с более широко распределенным поверхностным зарядом способен с привлечением электростатических взаимодействий связывать также олигомеры и протофибриллы Aβ. Электростатическое притяжение также препятствует образованию структурированных зародышей, участвующих в фибриллообразовании.
В то же время некоторые биохимические модификации ЧСА, вызванные окислительным и нитрозативным стрессом, сопровождающим БА, такие как нитрование остатков тирозина и гликирование, напротив, демонстрируют меньший ингибирующий эффект по сравнению с немодифицированным альбумином, как предполагается, в связи с тем, что преимущественно связывают Aβ уже после его конформационной перестройки, и выступают, таким образом, зародышами фибриллообразования (Ramos-Fernández et al., 2014).
Эти данные поддерживают модель “мономер-стабилизатор”, которая объясняет ингибирующее действие ЧСА тем, что он стабилизирует вторичную структуру Aβ и блокирует его конформационную перестройку, необходимую для запуска следующей стадии – зародышеобразования.
В отличие от мономера, во взаимодействии с олигомерами Aβ участвуют три сайта ЧСА (Milojevic et al., 2007, 2009; Milojevic, Melacini, 2011; Zhao, Guo, 2021). Высокая аффинность сайтов приводит к тому, что ЧСА способен эффективно блокировать их дальнейший рост по механизму “мономер-конкурент”, связываясь с преимущественно неупорядоченным С-концом растущей протофибриллы (Algamal et al., 2017). Этот механизм особенно важен в цереброспинальной жидкости, где концентрация ЧСА недостаточна для полного связывания мономерного Aβ (Stanyon, Viles, 2012). Однако в плазме, где концентрация ЧСА выше, чем в цереброспинальной жидкости, связывание мономера также играет важную роль в механизме влияния ЧСА на фибриллогенез (Milojevic et al., 2009; Reyes Barcelo et al., 2009).
Таким образом, исследования подтверждают регуляцию ЧСА всех стадий фибриллообразования (Wang et al., 2016; Zhao, Guo, 2021). Преимущественное влияние того или иного механизма in vivo в зависимости от локализации в организме, сопутствующих процессов и стадии развития БА еще предстоит установить.
РАЗРАБОТКА НОВЫХ ПОДХОДОВ К ТЕРАПИИ БОЛЕЗНИ АЛЬЦГЕЙМЕРА, ОСНОВАННЫХ НА ВЗАИМОДЕЙСТВИИ ЧЕЛОВЕЧЕСКОГО СЫВОРОТОЧНОГО АЛЬБУМИНА С Aβ
Первый терапевтический подход к лечению БА, направленный на ускорение выведения Aβ из ЦНС пациентов и основанный на замещении плазмы пациента препаратом ЧСА (5% раствор) производства фирмы Grifols (Испания), был предложен Мерсе Боада с соавторами (Boada et al., 2009) (рис. 2 ). Исследователи выдвинули гипотезу, что замена ЧСА пациента на очищенный альбумин позволит ускорить транспорт Aβ через гематоэнцефалический барьер, сдвинув равновесие между ЦНС и периферическим кровотоком в сторону последнего.
Рис. 2.
Мерсе Боада с соавторами впервые предложили метод для лечения болезни Альцгеймера, основанный на замещении плазмы пациента раствором 5% сывороточного альбумина производства фирмы Grifols (Испания). С помощью процедуры плазмафереза из кровотока вместе с плазмой удаляется собственный ЧСА пациента в комплексе с Aβ. Взамен в кровоток вводится фармакологический препарат альбумина, не связанного с Aβ, что позволяет ускорить выведение Aβ из ЦНС.
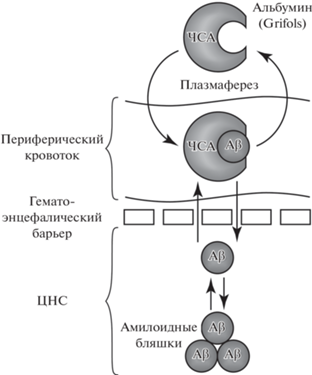
В 2009 г. были опубликованы первые данные клинических исследований, проходивших в три этапа (Boada et al., 2009). Первые два этапа представляли собой пилотное исследование нового терапевтического подхода с участием семи пациентов с установленным диагнозом БА. Пациентам дважды в неделю на протяжении трех недель проводили плазмаферез с заменой плазмы на очищенный фармакологический препарат сывороточного альбумина. Мониторинг состояния пациентов проводили в течение года. В ходе исследования были отмечены “пилообразные” колебания концентрации Aβ в образцах плазмы, строго ассоциированные по времени с проведением процедур, что в совокупности со стабилизацией когнитивного статуса пациентов было интерпретировано исследователями как усиление клиренса Aβ из ЦНС. Результаты второй фазы клинических испытаний (третий этап), в которую были включены 42 пациента с диагнозом БА, подтвердили результаты пилотных исследований и показали, что пациенты, получавшие лечение, обладали лучшими показателями когнитивного статуса по сравнению с контрольной группой (Boada et al., 2009, 2017). Кроме того, были получены данные о влиянии процедуры плазмафереза с заменой альбумина на уровень Aβ в спинномозговой жидкости и плазме пациентов и результаты нейропсихологических тестов на внимание и языковую функцию (Boada et al., 2017). Прохождение курса плазмафереза способствовало увеличению концентрации Aβ42 в спинномозговой жидкости пациентов и статистически значимому снижению его уровня в плазме, что указывало, по мнению авторов, на модуляцию метаболизма Aβ42 проводимым лечением. Наряду с устойчивой тенденцией к стабилизации показателей когнитивного статуса по сравнению с контрольной группой было отмечено статистически значимое улучшение памяти и языковой функции пациентов, прошедших лечение. В то же время авторы отмечают ухудшение поведенческих и функциональных показателей у пациентов, подвергшихся процедуре плазмафереза, по сравнению с контролем. У многих пациентов плазмаферез был связан с обострением психиатрической симптоматики, в том числе с повышенным уровнем тревожности. Описанные негативные эффекты полностью проходили после окончания активной фазы лечения. Временный характер негативных эффектов, стабилизация когнитивных показателей, а также устойчивое улучшение памяти и языковой функции позволили считать предложенный терапевтический подход перспективным (Boada et al., 2017).
В 2012 г. стартовала 2b/3 фаза клинических испытаний, названная AMBAR (Alzheimer’s Management By Albumin Replacement, EudraCT#: 2011–001598-25; ClinicalTrials.gov ID: NCT01561053). В исследовании приняли участие 347 пациентов, которые были разделены на четыре группы. Три группы получали лечение в виде курса плазмафереза с использованием фармакологического препарата ЧСА (5 или 20% раствора, Grifols), в двух из которых данный препарат комбинировали с препаратом иммуноглобулина (Флебогамма 5% ДИФ, Grifols) (Boada et al., 2020). Контрольная группа получала плацебо. Согласно данным, полученным с помощью шкалы оценки БА – когнитивной субшкалы (Disease Assessment Scale – Cognitive Subscale, ADAS-Cog) и шкалы оценки повседневной деятельности (Alzheimer’s Disease Cooperative Study – Activities of Daily Living, ADCS-ADL), проводимая терапия позволила значимо замедлить когнитивные и функциональные ухудшения у пациентов с БА средней тяжести. Исследователи также проводили общую оценку состояния пациентов с использованием шкалы оценки тяжести деменции (Clinical Dementia Rating – Sum of Boxes scale, CDR-sb) и шкалы общей клинической оценки (Alzheimer’s Disease Cooperative Study – Clinical Global Impression of Change, ADCS-CGIC). Результаты данных исследований показали положительные эффекты проводимой терапии: для пациентов с БА средней тяжести было отмечено замедление развития симптомов БА по сравнению с контрольной группой, а для пациентов с легкой формой заболевания были отмечены значимые улучшения в сравнении с их состоянием до проведения терапии (Boada et al., 2020). При анализе биохимических показателей спинномозговой жидкости у пациентов с БА средней степени тяжести, прошедших курс терапии, были выявлены стабилизация уровня Aβ42 и более низкое содержание общего и фосфорилированного тау-белка по сравнению с контрольной группой (Boada et al., 2020). Поскольку прогрессию БА связывают со снижением уровня Aβ42 (Andreasen et al., 1999) и повышением уровня тау-белка (Sjogren, 2001) в спинномозговой жидкости пациентов, то полученные результаты могут свидетельствовать о снижении содержания основных участников патогенеза БА в ЦНС (Loeffler, 2020). В то же время для оценки изменения количества отложений Aβ и тау-белка в головном мозге пациентов в будущем необходимо получить данные позитронно-эмиссионной томографии (Loeffler, 2020).
В дальнейшем были предложены терапевтические подходы, основанные на введении препаратов ЧСА непосредственно в ЦНС. Положительной стороной такого решения является отсутствие гематоэнцефалического барьера, что предполагает возможность диффузии введенного препарата непосредственно в интерстициальное пространство головного мозга. Эзра с соавторами (Ezra et al., 2016) провели исследование действия ЧСА на амилоидные отложения в головном мозге трансгенных (3xTg-AD) мышей при его интрацеребровентрикулярном введении. Наряду с уменьшением содержания Aβ в головном мозге мышей, исследователи наблюдали снижение уровня тау-белка и увеличение стабильности микротрубочек. Более того, у животных отмечалось снижение выраженности воспалительных процессов и восстановление гематоэнцефалического барьера (Ezra et al., 2016). Накопленные данные клинических испытаний и исследований на животных моделях, подтверждающих положительный эффект ЧСА на течение БА, стали основой для альтернативного терапевтического подхода, описанного в работе Менендез-Гонзалез и Гаспарович (Menendez-Gonzalez, Gasparovic, 2019). Исследователи предложили использовать метод ликворофереза с заменой ЧСА на фармакологический препарат альбумина. Данный подход, несомненно, имеет преимущества, поскольку, как уже отмечалось, позволяет обойти гематоэнцефалический барьер и не зависит от метаболизма Aβ на периферии (вне ЦНС). В то же время метод ликворофереза не имеет такого широкого распространения в сравнении с плазмаферезом, а введение препаратов альбумина в субарахноидальное пространство в клинической практике не проводилось (Menendez-Gonzalez, Gasparovic, 2019). Пока данный терапевтический подход требует серьезной доработки.
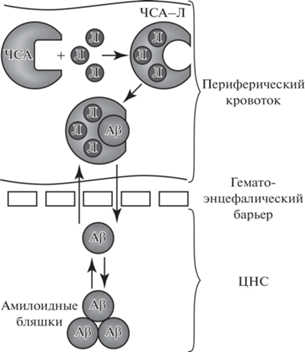
Другим перспективным направлением разработки новых терапевтических и профилактических подходов для БА является поиск модификаций молекулы ЧСА, усиливающих его сродство к Aβ. Ишима с соавторами (Ishima et al., 2020) предложили набор мутантных форм 2-го домена ЧСА, обладающих повышенным сродством к Aβ. По мнению авторов, полученные мутантные формы способны эффективнее по сравнению с диким типом сдвигать равновесие между ЦНС и периферическим кровотоком, усиливая выведение Aβ из головного мозга пациентов. Ван c соавторами (Wang et al., 2019) показали, что подобного эффекта можно добиться, использовав основную форму ЧСА, в структуре которого часть карбоксильных групп заменены на аминогруппы. Внедрение модифицированного ЧСА в клиническую практику требует прохождения полного цикла доклинических и клинических исследований. В то же время сродство ЧСА к Aβ можно модулировать с помощью лигандов ЧСА, многие из которых уже используются в виде пищевых добавок или лекарственных препаратов.
ВЛИЯНИЕ ЛИГАНДОВ ЧЕЛОВЕЧЕСКОГО СЫВОРОТОЧНОГО АЛЬБУМИНА НА ЕГО ВЗАИМОДЕЙСТВИЕ С Aβ
ЧСА обладает уникальной способностью связывать широкий спектр эндогенных и экзогенных веществ (Kragh-Hansen, 1990; Fasano et al., 2005). Взаимодействие лигандов с сывороточным альбумином влияет на их фармакокинетические и фармакодинамические свойства (Ishima et al., 2020). В то же время сами лиганды способны оказывать влияние на структуру и функциональные свойства ЧСА, в том числе модулировать его взаимодействие с Aβ.
К настоящему моменту накоплены данные о существовании группы органических веществ-лигандов ЧСА экзогенного и эндогенного происхождения, способных усиливать сродство ЧСА к Aβ. В исследованиях in vitro показано, что нейромедиатор серотонин повышает сродство ЧСА к Aβ40 и Aβ42 в 7–17 раз (Litus et al., 2021). Эти данные согласуются с результатами эпидемиологических исследований о наличии дегенеративных изменений серотонинергической системы (Hirao, Smith, 2014) и снижении уровня серотонина в биологических жидкостях (Whiley et al., 2021) у пациентов с БА, а также о сокращении содержания Aβ в головном мозге возрастных пациентов при приеме ингибиторов обратного захвата серотонина (Cirrito et al., 2011). Вместе с тем концентрация серотонина, необходимая для увеличения сродства ЧСА к Aβ, достигает 1 мМ (Litus et al., 2021), что в физиологических условиях может наблюдаться только локально, в просвете синаптической щели (Bunin, Wightman, 1998), но недостижимо в периферическом кровотоке. В этой связи возможность прямого использования серотонина в качестве лекарственного средства, усиливающего сродство ЧСА к Aβ с целью терапии или профилактики БА, требует дальнейшего изучения.
ЧСА является основным переносчиком практически нерастворимых в воде жирных кислот (ЖК) (Vusse, 2009) и участвует в транспорте холестерина (Zhao, Marcel, 1996). При этом связывание мажорных для плазмы крови ЖК (линолевая (ЛК), арахидоновая (АК), пальмитиновая (ПК)) влияет на сродство ЧСА к Aβ и его способность ингибировать образование фибрилл. Показано, что связывание ЛК и АК способствует взаимодействию ЧСА с Aβ42 (константа диссоциации комплекса в их присутствии снижается в 2.2–2.6 раз) (Litus et al., 2019). В этой связи наблюдаемое 6-кратное снижение уровня ЛК в плазме крови пациентов с БА (Cunnane et al., 2012) может приводить к значимому снижению сродства ЧСА к Aβ42 и накоплению последнего в ЦНС пациентов. Также к фактам, подтверждающим участие ЛК в патогенезе БА, можно отнести снижение риска развития БА при потреблении полиненасыщенных ЖК (Morris et al., 2003; Laitinen et al., 2006). В то же время потребление в пищу насыщенных ЖК и холестерина прямо коррелирует с риском развития БА (Morris et al., 2003; Laitinen et al., 2006). Это, в свою очередь, может являться следствием подавления способности ЧСА ингибировать образование Aβ фибрилл при связывании данных лигандов (Bode et al., 2018). При этом и насыщенные ЖК, и холестерин могут конкурировать с Aβ за связывание с ЧСА, либо их связывание с альбумином приводит к структурным изменениям белка, разрушающим центр связывания Aβ (Algamal et al., 2013).
Помимо низкомолекулярных органических соединений, ЧСА участвует в транспорте двухвалентных ионов металлов (Fasano et al., 2005). В частности, 15% общего содержания Сu2+ в крови связано с ЧСА (Bal et al., 2013). Несмотря на наличие нескольких сайтов связывания, в физиологических условиях Сu2+ заполнен только N-концевой сайт ЧСА. Многие исследования свидетельствуют о взаимосвязи между нарушением гомеостаза Сu2+ и развитием БА (Bagheri et al., 2017). При этом ЧСА способен выполнять функцию хелатора Сu2+, предотвращая индуцированную данным ионом агрегацию Aβ (Choi et al., 2017). С другой стороны, связывание Сu2+ снижает кинетическую константу диссоциации комплекса ЧСА с Aβ на 31–37% (Litus et al., 2019), что указывает на возможность усиления взаимодействия ЧСА с Aβ в присутствии двухвалентных ионов металлов.
В контексте разработки новых подходов для лечения и профилактики БА среди экзогенных лигандов ЧСА особый интерес представляют широко используемые безрецептурные лекарственные препараты. По данным последних исследований in vitro, ибупрофен в терапевтических концентрациях увеличивает сродство ЧСА к мономерным формам Aβ40 и Aβ42 в 3–5 раз и усиливает ингибирующий эффект ЧСА по отношению к процессу образования фибрилл Aβ (Litus et al., 2022). В этом случае данные in vitro подкрепляются более ранними наблюдениями in vivo, в которых отмечалось снижение риска развития БА у пациентов, длительно принимающих нестероидные противовоспалительные препараты, в том числе и ибупрофен (Wang et al., 2015). Данные, полученные на животных моделях, подтверждают снижение содержания отложений Aβ и гиперфосфорелированного тау-белка, подавление воспалительных процессов в головном мозге, а также уменьшение выраженности когнитивных нарушений под действием ибупрофена (Lim et al., 2000; Yan et al., 2003; McKee et al., 2008). Однако данные клинических исследованиях об эффективности нестероидных противовоспалительных препаратов для лечения БА, как уже отмечалось, неоднозначны (Hayden et al., 2007; Tschanz et al., 2013; Miguel-Álvarez et al., 2015; Ali et al., 2019), поэтому возможность использования ибупрофена для лечения и профилактики БА требует дальнейшего изучения.
К низкомолекулярным лигандам ЧСА, препятствующим его взаимодействию с Aβ, относятся толбутамид и варфарин. Низкомолекулярный антидиабетический препарат толбутамид в терапевтических концентрациях усиливает образование амилоидных фибрилл в присутствии, но не в отсутствии ЧСА (Bohrmann et al., 1999). Авторы предполагают, что толбутамид конкурирует с Aβ за связывание с одним и тем же участком ЧСА, вытесняя его из альбумина и увеличивая свободную концентрацию Aβ. Снижение способности ЧСА ингибировать реакцию образования фибрилл Aβ наблюдается и в присутствии варфарина (Bode et al., 2018). Стоит отметить, что указанные эффекты изучались только в in vitro экспериментах. Могут ли толбутамид или варфарин изменять сродство ЧСА к Aβ in vivo в физиологических или патологических условиях, в частности может ли назначение данных лекарственных препаратов приводить к накоплению Aβ в головном мозге пациентов, – вопросы, требующие дальнейших исследований.
В целом, усиление сродства ЧСА к Aβ под влиянием лигандов сывороточного альбумина может в будущем стать основой для разработки новой группы лекарственных препаратов, использование которых будет ускорять выведение или препятствовать накоплению Aβ в головном мозге пациентов с установленным диагнозом БА, а также имеющих высокий риск развития данного заболевания (рис. 3 ).
ЗАКЛЮЧЕНИЕ
Несмотря на большое количество исследований, посвященных изучению БА и разработке методов ее лечения и профилактики, до сих пор представления о ее патогенезе остаются фрагментарными, а большая часть лекарственных препаратов, применяемых в широкой клинической практике, облегчают симптомы, но не влияют на развитие заболевания. Новым направлением в лечении БА является снижение содержания Aβ в головном мозге пациентов путем сдвига равновесия “ЦНС–периферический кровоток” в сторону последнего. ЧСА, как основной переносчик Aβ в крови, стал мишенью для разработки новых терапевтических подходов. Среди предложенных в литературе методов лечения, использующих ЧСА, можно выделить: 1) введение фармакологического препарата ЧСА в кровь пациента; 2) введение фармакологического препарата ЧСА в ЦНС пациента; 3) использование модифицированных форм ЧСА, обладающих повышенным сродством к Aβ; 4) усиление взаимодействия ЧСА–Aβ путем воздействия лигандов ЧСА. Использование плазмафереза с заменой ЧСА уже было апробировано в клинической практике и показало положительные результаты. В то же время нельзя не отметить влияние такого подхода на метаболизм множества лигандов ЧСА, которые удаляются из кровотока пациента вместе с их белком-переносчиком. Безусловно, такие методы, как прямое введение ЧСА в ЦНС (интрацеребровентрикулярно или субарахноидально) и использование модифицированных форм ЧСА, требуют проведения дополнительных исследований in vitro и на животных моделях и далеки от внедрения в клиническую практику, в то время как лиганды ЧСА являются уже одобренными для использования и широко известными лекарственными веществами или естественными метаболитами, что существенно упрощает разработку новых подходов к лечению и профилактике БА с их использованием. Необходимо отметить, что разработка любого из предложенных подходов даст толчок для лучшего понимания роли Aβ в развитии БА.
Список литературы
Algamal M., Milojevic J., Jafari N., Zhang W., Melacini G., 2013. Mapping the interactions between the Alzheimer’s Aβ-peptide and human serum albumin beyond domain resolution // Biophys. J. V. 105. № 7. P. 1700–1709. https://doi.org/10.1016/j.bpj.2013.08.025
Algamal M., Ahmed R., Jafari N., Ahsan B., Ortega J., Melacini G., 2017. Atomic-resolution map of the interactions between an amyloid inhibitor protein and amyloid β (Aβ) peptides in the monomer and protofibril states // J. Biol. Chem. V. 292. № 42. P. 17158–17168. https://doi.org/10.1074/jbc.M117.792853
Ali M.M., Ghouri R.G., Ans A.H., Akbar A., Toheed A., 2019. Recommendations for anti-inflammatory treatments in Alzheimer’s disease: A comprehensive review of the literature // Cureus. V. 11. № 5. Art. e4620. https://doi.org/10.7759/cureus.4620
Alonso A.C., Zaidi T., Grundke-Iqbal I., Iqbal K., 1994. Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease // Proc. Natl. Acad. Sci. V. 91. № 12. P. 5562–5566. https://doi.org/10.1073/pnas.91.12.5562
Andreasen N., Hesse C., Davidsson P., Minthon L., Wallin A. et al., 1999. Cerebrospinal fluid beta-amyloid 1-42 in Alzheimer disease: differences between early- and late-onset Alzheimer disease and stability during the course of disease // Arch. Neurol. V. 56. № 6. P. 673–680. https://doi.org/10.1001/archneur.56.6.673
Arvanitakis Z., Shah R.C., Bennett D.A., 2019. Diagnosis and management of dementia: Review // JAMA. V. 322. № 16. P. 1589–1599. https://doi.org/10.1001/jama.2019.4782
Azizi G., Navabi S.S., Al-Shukaili A., Seyedzadeh M.H., Yazdani R., Mirshafiey A., 2015. The role of inflammatory mediators in the pathogenesis of Alzheimer’s disease // Sultan Qaboos Univ. Med. J. V. 15. № 3. P. e305–316. https://doi.org/10.18295/squmj.2015.15.03.002
Bagheri S., Squitti R., Haertlé T., Siotto M., Saboury A.A., 2017. Role of copper in the onset of Alzheimer’s disease compared to other metals // Front. Aging Neurosci. V. 9. Art. 446. https://doi.org/10.3389/fnagi.2017.00446
Bal W., Sokołowska M., Kurowska E., Faller P., 2013. Binding of transition metal ions to albumin: sites, affinities and rates // Biochim. Biophys. Acta. V. 1830. № 12. P. 5444–5455. https://doi.org/10.1016/j.bbagen.2013.06.018
Bali J., Gheinani A.H., Zurbriggen S., Rajendran L., 2012. Role of genes linked to sporadic Alzheimer’s disease risk in the production of β-amyloid peptides // Proc. Natl. Acad. Sci. USA. V. 109. № 38. P. 15307–15311. https://doi.org/10.1073/pnas.1201632109
Baumketner A., Bernstein S.L., Wyttenbach T., Bitan G., Teplow D.B. et al., 2006. Amyloid beta-protein monomer structure: a computational and experimental study // Protein Sci. V. 15. № 3. P. 420–428. https://doi.org/10.1110/ps.051762406
Bernstein S.L., Wyttenbach T., Baumketner A., Shea J.-E., Bitan G. et al., 2005. Amyloid β-Protein: Monomer structure and early aggregation states of Aβ42 and its Pro 19 alloform // J. Am. Chem. Soc. V. 127. № 7. P. 2075–2084. https://doi.org/10.1021/ja044531p
Biere A.L., Ostaszewski B., Stimson E.R., Hyman B.T., Maggio J.E., Selkoe D.J., 1996. Amyloid β-Peptide is transported on lipoproteins and albumin in human plasma // J. Biol. Chem. V. 271. № 51. P. 32916–32922. https://doi.org/10.1074/jbc.271.51.32916
Bitan G., Kirkitadze M.D., Lomakin A., Vollers S.S., Benedek G.B., Teplow D.B., 2003. Amyloid beta-protein (Abeta) assembly: Abeta 40 and Abeta 42 oligomerize through distinct pathways // Proc. Natl. Acad. Sci. USA. V. 100. № 1. P. 330–335. https://doi.org/10.1073/pnas.222681699
Boada M., Ortiz P., Anaya F., Hernández I., Muñoz J. et al., 2009. Amyloid-targeted therapeutics in Alzheimer’s disease: Use of human albumin in plasma exchange as a novel approach for Abeta mobilization // Drug News Perspect. V. 22. № 6. P. 325–339. https://doi.org/10.1358/dnp.2009.22.6.1395256
Boada M., Anaya F., Ortiz P., Olazarán J., Shua-Haim J.R. et al., 2017. Efficacy and safety of plasma exchange with 5% albumin to modify cerebrospinal fluid and plasma amyloid-β concentrations and cognition outcomes in Alzheimer’s disease patients: Amulticenter, randomized, controlled clinical trial // J. Alzheimers Dis. V. 56. № 1. P. 129–143. https://doi.org/10.3233/JAD-160565
Boada M., López O.L., Olazarán J., Núñez L., Pfeffer M. et al., 2020. A randomized, controlled clinical trial of plasma exchange with albumin replacement for Alzheimer’s disease: Primary results of the AMBAR Study // Alzheimers Dement. V. 16. № 10. P. 1412–1425. https://doi.org/10.1002/alz.12137
Bode D.C., Stanyon H.F., Hirani T., Baker M.D., Nield J., Viles J.H., 2018. Serum albumin’s protective inhibition of amyloid-β fiber formation is suppressed by cholesterol, fatty acids and warfarin // J. Mol. Biol. V. 430. № 7. P. 919–934. https://doi.org/10.1016/j.jmb.2018.01.008
Bohrmann B., Tjernberg L., Kuner P., Poli S., Levet-Trafit B. et al., 1999. Endogenous proteins controlling amyloid beta-peptide polymerization. Possible implications for beta-amyloid formation in the central nervous system and in peripheral tissues // J. Biol. Chem. V. 274. № 23. P. 15990–15995. https://doi.org/10.1074/jbc.274.23.15990
Brier M.R., Gordon B., Friedrichsen K., McCarthy J., Stern A. et al., 2016. Tau and Aβ imaging, CSF measures, and cognition in Alzheimer’s disease // Sci. Transl. Med. V. 8. № 338. Art. 338ra66. https://doi.org/10.1126/scitranslmed.aaf2362
Brinkman S.D., Gershon S., 1983. Measurement of cholinergic drug effects on memory in alzheimer’s disease // Neurobiol. Aging. V. 4. № 2. P. 139–145. https://doi.org/10.1016/0197-4580(83)90038-6
Bunin M.A., Wightman R.M., 1998. Quantitative evaluation of 5-hydroxytryptamine (serotonin) neuronal release and uptake: An investigation of extrasynaptic transmission // J. Neurosci. V. 18. № 13. P. 4854–4860. https://doi.org/10.1523/JNEUROSCI.18-13-04854.1998
Butterfield D.A., Lauderback C.M., 2002. Lipid peroxidation and protein oxidation in Alzheimer’s disease brain: Potential causes and consequences involving amyloid β-peptide-associated free radical oxidative stress // Free Radic. Biol. Med. V. 32. № 11. P. 1050–1060. https://doi.org/10.1016/S0891-5849(02)00794-3
Butterfield D.A., Reed T., Newman S.F., Sultana R., 2007. Roles of amyloid β-peptide-associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer’s disease and mild cognitive impairment // Free Radic. Biol. Med. V. 43. № 5. P. 658–677. https://doi.org/10.1016/j.freeradbiomed.2007.05.037
Carrillo-Mora P., Luna R., Colín-Barenque L., 2014. Amyloid beta: Multiple mechanisms of toxicity and only some protective effects? // Oxid. Med. Cell. Longev. V. 2014. Art. 795375. https://doi.org/10.1155/2014/795375
Cheignon C., Tomas M., Bonnefont-Rousselot D., Faller P., Hureau C., Collin F., 2018. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease // Redox Biol. V. 14. P. 450–464. https://doi.org/10.1016/j.redox.2017.10.014
Choi T.S., Lee H.J., Han J.Y., Lim M.H., Kim H.I., 2017. Molecular insights into human serum albumin as a receptor of amyloid-β in the extracellular region // J. Am. Chem. Soc. V. 139. № 43. P. 15437–15445. https://doi.org/10.1021/jacs.7b08584
Christen Y., 2000. Oxidative stress and Alzheimer disease // Am. J. Clin. Nutr. V. 71. № 2. P. 621S–629S. https://doi.org/10.1093/ajcn/71.2.621s
Cirrito J.R., Disabato B.M., Restivo J.L., Verges D.K., Goebel W.D. et al., 2011. Serotonin signaling is associated with lower amyloid-β levels and plaques in transgenic mice and humans // Proc. Natl. Acad. Sci. USA. V. 108. № 36. P. 14968–14973. https://doi.org/10.1073/pnas.1107411108
Costa M., Ortiz A.M., Jorquera J.I., 2012. Therapeutic albumin binding to remove amyloid-β // J. Alzheimers Dis. V. 29. № 1. P. 159–170. https://doi.org/10.3233/JAD-2012-111139
Cuberas-Borrós G., Roca I., Boada M., Tárraga L., Hernández I. et al., 2018. Longitudinal neuroimaging analysis in mild-moderate Alzheimer’s disease patients treated with plasma exchange with 5% human albumin // J. Alzheimers Dis. V. 61. № 1. P. 321–332. https://doi.org/10.3233/JAD-170693
Cunnane S.C., Schneider J.A., Tangney C., Tremblay-Mercier J., Fortier M. et al., 2012. Plasma and brain fatty acid profiles in mild cognitive impairment and Alzheimer’s disease // J. Alzheimers Dis. V. 29. № 3. P. 691–697. https://doi.org/10.3233/JAD-2012-110629
Deane R., Bell R.D., Sagare A., Zlokovic B.V., 2009. Clearance of amyloid-beta peptide across the blood-brain barrier: Implication for therapies in Alzheimer’s disease // CNS Neurol. Disord. Drug Targets. V. 8. № 1. P. 16–30. https://doi.org/10.2174/187152709787601867
DeMattos R.B., Bales K.R., Parsadanian M., O’Dell M.A., Foss E.M. et al., 2002. Plaque-associated disruption of CSF and plasma amyloid-beta (Abeta) equilibrium in a mouse model of Alzheimer’s disease // J. Neurochem. V. 81. № 2. P. 229–236. https://doi.org/10.1046/j.1471-4159.2002.00889.x
Du X., Wang X., Geng M., 2018. Alzheimer’s disease hypothesis and related therapies // Transl. Neurodegener. V. 7. № 1. Art. 2. https://doi.org/10.1186/s40035-018-0107-y
Ezra A., Rabinovich-Nikitin I., Rabinovich-Toidman P., Solomon B., 2016. Multifunctional effect of human serum albumin reduces Alzheimer’s disease related pathologies in the 3xTg mouse model // J. Alzheimers Dis. V. 50. № 1. P. 175–188. https://doi.org/10.3233/JAD-150694
Fändrich M., 2007. On the structural definition of amyloid fibrils and other polypeptide aggregates // Cell. Mol. Life Sci. V. 64. № 16. P. 2066–2078. https://doi.org/10.1007/s00018-007-7110-2
Fändrich M., Meinhardt J., Grigorieff N., 2009. Structural polymorphism of Alzheimer Aβ and other amyloid fibrils // Prion. V. 3. № 2. P. 89–93. https://doi.org/10.4161/pri.3.2.8859
Fasano M., Curry S., Terreno E., Galliano M., Fanali G., et al., 2005. The extraordinary ligand binding properties of human serum albumin // IUBMB Life. V. 57. № 12. P. 787–796. https://doi.org/10.1080/15216540500404093
GBD 2019 Dementia Forecasting Collaborators, 2022. Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: An analysis for the Global Burden of Disease Study 2019 // Lancet. Public Heal. V. 7. № 2. P. e105–e125. https://doi.org/10.1016/S2468-2667(21)00249-8
Gella A., Durany N., 2009. Oxidative stress in Alzheimer disease // Cell Adh. Migr. V. 3. № 1. P. 88–93. https://doi.org/10.4161/cam.3.1.7402
Ghersi-Egea J.F., Gorevic P.D., Ghiso J., Frangione B., Patlak C.S., Fenstermacher J.D., 1996. Fate of cerebrospinal fluid-borne amyloid beta-peptide: Rapid clearance into blood and appreciable accumulation by cerebral arteries // J. Neurochem. V. 67. № 2. P. 880–883. https://doi.org/10.1046/j.1471-4159.1996.67020880.x
Gibson G.L., Allsop D., Austen B.M., 2004. Induction of cellular oxidative stress by the beta-amyloid peptide involved in Alzheimer’s disease // Protein Pept. Lett. V. 11. № 3. P. 257–270. https://doi.org/10.2174/0929866043407101
Goedert M., Spillantini M.G., 2001. Tau gene mutations and neurodegeneration // Biochem. Soc. Symp. V. 67. № 67. P. 59–71. https://doi.org/10.1042/bss0670059
Gong Y., Chang L., Viola K.L., Lacor P.N., Lambert M.P. et al., 2003. Alzheimer’s disease-affected brain: Presence of oligomeric A beta ligands (ADDLs) suggests a molecular basis for reversible memory loss // Proc. Natl. Acad. Sci. USA. V. 100. № 18. P. 10417–10422. https://doi.org/10.1073/pnas.1834302100
Hayden K.M., Zandi P.P., Khachaturian A.S., Szekely C.A., Fotuhi M. et al., 2007. Does NSAID use modify cognitive trajectories in the elderly? The Cache County Study // Neurology. V. 69. № 3. P. 275–282. https://doi.org/10.1212/01.wnl.0000265223.25679.2a
Hirao K., Smith G.S., 2014. Positron emission tomography molecular imaging in late-life depression // J. Geriatr. Psychiatry Neurol. V. 27. № 1. P. 13–23. https://doi.org/10.1177/0891988713516540
Ishima Y., Mimono A., Tuan Giam Chuang V., Fukuda T., Kusumoto K. et al., 2020. Albumin domain mutants with enhanced Aβ binding capacity identified by phage display analysis for application in various peripheral Aβ elimination approaches of Alzheimer’s disease treatment // IUBMB Life. V. 72. № 4. P. 641–651. https://doi.org/10.1002/iub.2203
Kayed R., Head E., Thompson J.L., McIntire T.M., Milton S.C. et al., 2003. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis // Science. V. 300. № 5618. P. 486–489. https://doi.org/10.1126/science.1079469
Kinney J.W., Bemiller S.M., Murtishaw A.S., Leisgang A.M., Salazar A.M., Lamb B.T., 2018. Inflammation as a central mechanism in Alzheimer’s disease // Alzheimers Dement. Transl. Res. Clin. Interv. V. 4. № 1. P. 575–590. https://doi.org/10.1016/j.trci.2018.06.014
Kirkitadze M.D., Condron M.M., Teplow D.B., 2001. Identification and characterization of key kinetic intermediates in amyloid beta-protein fibrillogenesis // J. Mol. Biol. V. 312. № 5. P. 1103–1119. https://doi.org/10.1006/jmbi.2001.4970
Kirschner D.A., Abraham C., Selkoe D.J., 1986. X-ray diffraction from intraneuronal paired helical filaments and extraneuronal amyloid fibers in Alzheimer disease indicates cross-beta conformation // Proc. Natl. Acad. Sci. USA. V. 83. № 2. P. 503–507. https://doi.org/10.1073/pnas.83.2.503
Kragh-Hansen U., 1990. Structure and ligand binding properties of human serum albumin // Dan. Med. Bull. V. 37. № 1. P. 57–84. http://www.ncbi.nlm.nih.gov/pubmed/2155760
Kumar A., Sidhu J., Goyal A., Tsao J.W., 2022. Alzheimer Disease. Treasure Island (FL): StatPearls Publishing. http://www.ncbi.nlm.nih.gov/pubmed/29763097
Kuo Y.M., Kokjohn T.A., Kalback W., Luehrs D., Galasko D.R. et al., 2000. Amyloid-beta peptides interact with plasma proteins and erythrocytes: Implications for their quantitation in plasma // Biochem. Biophys. Res. Commun. V. 268. № 3. P. 750–756. https://doi.org/10.1006/bbrc.2000.2222
Laitinen M.H., Ngandu T., Rovio S., Helkala E.-L., Uusitalo U. et al., 2006. Fat intake at midlife and risk of dementia and Alzheimer’s disease: A population-based study // Dement. Geriatr. Cogn. Disord. V. 22. № 1. P. 99–107. https://doi.org/10.1159/000093478
Lim G.P., Yang F., Chu T., Chen P., Beech W. et al., 2000. Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer’s disease // J. Neurosci. V. 20. № 15. P. 5709–5714. https://doi.org/10.1523/JNEUROSCI.20-15-05709.2000
Litus E.A., Kazakov A.S., Sokolov A.S., Nemashkalova E.L., Galushko E.I. et al., 2019. The binding of monomeric amyloid β peptide to serum albumin is affected by major plasma unsaturated fatty acids // Biochem. Biophys. Res. Commun. V. 510. № 2. P. 248–253. https://doi.org/10.1016/j.bbrc.2019.01.081
Litus E.A., Kazakov A.S., Deryusheva E.I., Nemashkalo-va E.L., Shevelyova M.P. et al., 2021. Serotonin promotes serum albumin interaction with the monomeric amyloid-β peptide // Int. J. Mol. Sci. V. 22. № 11. Art. 5896. https://doi.org/10.3390/ijms22115896
Litus E.A., Kazakov A.S., Deryusheva E.I., Nemashkalova E.L., Shevelyova M.P. et al., 2022. Ibuprofen favors binding of amyloid-β peptide to its depot, serum albumin // Int. J. Mol. Sci. V. 23. № 11. Art. 6168. https://doi.org/10.3390/ijms23116168
Llewellyn J.D., Langa M.K., Friedland P.R., Lang A.I., 2010. Serum albumin concentration and cognitive impairment // Curr. Alzheimer Res. V. 7. № 1. P. 91–96. https://doi.org/10.2174/156720510790274392
Loeffler D.A., 2020. AMBAR, an encouraging Alzheimer’s trial that raises questions // Front. Neurol. V. 11. Art. 459. https://doi.org/10.3389/fneur.2020.00459
Matsuoka Y., Saito M., LaFrancois J., Saito M., Gaynor K. et al., 2003. Novel therapeutic approach for the treatment of Alzheimer’s disease by peripheral administration of agents with an affinity to β-amyloid // J. Neurosci. V. 23. № 1. P. 29–33. https://doi.org/10.1523/JNEUROSCI.23-01-00029.2003
McCormick J.W., Ammerman L., Chen G., Vogel P.D., Wise J.G., 2021. Transport of Alzheimer’s associated amyloid-β catalyzed by P-glycoprotein // PLoS One. V. 16. № 4. Art. e0250371. https://doi.org/10.1371/journal.pone.0250371
McKee A.C., Carreras I., Hossain L., Ryu H., Klein W.L. et al., 2008. Ibuprofen reduces Aβ, hyperphosphorylated tau and memory deficits in Alzheimer mice // Brain Res. V. 1207. P. 225–236. https://doi.org/10.1016/j.brainres.2008.01.095
Menendez-Gonzalez M., Gasparovic C., 2019. Albumin exchange in Alzheimer’s disease: Might CSF be an alternative route to plasma? // Front. Neurol. V. 10. Art. 1036. https://doi.org/10.3389/fneur.2019.01036
Meraz-Ríos M.A., Toral-Rios D., Franco-Bocanegra D., Villeda-Hernández J., Campos-Peña V., 2013. Inflammatory process in Alzheimer’s Disease // Front. Integr. Neurosci. V. 7. Art. 59. https://doi.org/10.3389/fnint.2013.00059
Metaxas A., Kempf S.J., 2016. Neurofibrillary tangles in Alzheimer’s disease: Elucidation of the molecular mechanism by immunohistochemistry and tau protein phospho-proteomics // Neural Regen. Res. V. 11. № 10. P. 1579–1581. https://doi.org/10.4103/1673-5374.193234
Miguel-Álvarez M., Santos-Lozano A., Sanchis-Gomar F., Fiuza-Luces C., Pareja-Galeano H. et al., 2015. Non-steroidal anti-inflammatory drugs as a treatment for Alzheimer’s disease: A systematic review and meta-analysis of treatment effect // Drugs Aging. V. 32. № 2. P. 139–147. https://doi.org/10.1007/s40266-015-0239-z
Milojevic J., Melacini G., 2011. Stoichiometry and affinity of the human serum albumin-Alzheimer’s Aβ peptide interactions // Biophys. J. V. 100. № 1. P. 183–192. https://doi.org/10.1016/j.bpj.2010.11.037
Milojevic J., Raditsis A., Melacini G., 2009. Human serum albumin inhibits Abeta fibrillization through a “monomer-competitor” mechanism // Biophys. J. V. 97. № 9. P. 2585–2594. https://doi.org/10.1016/j.bpj.2009.08.028
Milojevic J., Esposito V., Das R., Melacini G., 2007. Understanding the molecular basis for the inhibition of the Alzheimer’s Abeta-peptide oligomerization by human serum albumin using saturation transfer difference and off-resonance relaxation NMR spectroscopy // J. Am. Chem. Soc. V. 129. № 14. P. 4282–4290. https://doi.org/10.1021/ja067367+
Moreira P.I., Carvalho C., Zhu X., Smith M.A., Perry G., 2010. Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology // Biochim. Biophys. Acta. V. 1802. № 1. P. 2–10. https://doi.org/10.1016/j.bbadis.2009.10.006
Morris M.C., Evans D.A., Bienias J.L., Tangney C.C., Bennett D.A. et al., 2003. Dietary fats and the risk of incident Alzheimer disease // Arch. Neurol. V. 60. № 2. P. 194–200. https://doi.org/10.1001/archneur.60.2.194
Mullard A., 2021. Failure of first anti-tau antibody in Alzheimer disease highlights risks of history repeating // Nat. Rev. Drug Discov. V. 20. № 1. P. 3–5. https://doi.org/10.1038/d41573-020-00217-7
Murphy M.P., LeVine H., 2010. Alzheimer’s disease and the amyloid-β peptide // J. Alzheimers Dis. V. 19. № 1. P. 311–323. https://doi.org/10.3233/JAD-2010-1221
Pitschke M., Prior R., Haupt M., Riesner D., 1998. Detection of single amyloid beta-protein aggregates in the cerebrospinal fluid of Alzheimer’s patients by fluorescence correlation spectroscopy // Nat. Med. V. 4. № 7. P. 832–834. https://doi.org/10.1038/nm0798-832
Pizzino G., Irrera N., Cucinotta M., Pallio G., Mannino F. et al., 2017. Oxidative stress: Harms and benefits for human health // Oxid. Med. Cell. Longev. V. 2017. P. 1–13. https://doi.org/10.1155/2017/8416763
Poduslo J.F., Curran G.L., Sanyal B., Selkoe D.J., 1999. Receptor-mediated transport of human amyloid beta-protein 1-40 and 1-42 at the blood-brain barrier // Neurobiol. Dis. V. 6. № 3. P. 190–199. https://doi.org/10.1006/nbdi.1999.0238
Poorkaj P., Grossman M., Steinbart E., Payami H., Sadovnick A. et al., 2001. Frequency of tau gene mutations in familial and sporadic cases of non-Alzheimer dementia // Arch. Neurol. V. 58. № 3. P. 383–387. https://doi.org/10.1001/archneur.58.3.383
Qiang W., Yau W.-M., Luo Y., Mattson M.P., Tycko R., 2012. Antiparallel β-sheet architecture in Iowa-mutant β‑amyloid fibrils // Proc. Natl. Acad. Sci. V. 109. № 12. P. 4443–4448. https://doi.org/10.1073/pnas.1111305109
Ramos-Fernández E., Tajes M., Palomer E., Ill-Raga G., Bosch-Morató M. et al., 2014. Posttranslational nitro-glycative modifications of albumin in Alzheimer’s disease: Implications in cytotoxicity and amyloid-β peptide aggregation // J. Alzheimers Dis. V. 40. № 3. P. 643–657. https://doi.org/10.3233/JAD-130914
Rayner H.C., Hasking D.J., 1986. Hyperparathyroidism associated with severe hypercalcaemia and myocardial calcification despite minimal bone disease // BMJ. V. 293. № 6557. P. 1277–1278. https://doi.org/10.1136/bmj.293.6557.1277-a
Reyes Barcelo A.A., Gonzalez-Velasquez F.J., Moss M.A., 2009. Soluble aggregates of the amyloid-beta peptide are trapped by serum albumin to enhance amyloid-beta activation of endothelial cells // J. Biol. Eng. V. 3. № 1. Art. 5. https://doi.org/10.1186/1754-1611-3-5
Rivers-Auty J., Mather A.E., Peters R., Lawrence C.B., Brough D., 2020. Anti-inflammatories in Alzheimer’s disease – potential therapy or spurious correlate? // Brain Commun. V. 2. № 2. Art. fcaa109. https://doi.org/10.1093/braincomms/fcaa109
Roberts K.F., Elbert D.L., Kasten T.P., Patterson B.W., Sigurdson W.C. et al., 2014. Amyloid-β efflux from the central nervous system into the plasma // Ann. Neurol. V. 76. № 6. P. 837–844. https://doi.org/10.1002/ana.24270
Rodríguez-Martín T., Cuchillo-Ibáñez I., Noble W., Nyenya F., Anderton B.H., Hanger D.P., 2013. Tau phosphorylation affects its axonal transport and degradation // Neurobiol. Aging. V. 34. № 9. P. 2146–2157. https://doi.org/10.1016/j.neurobiolaging.2013.03.015
Rózga M., Kłoniecki M., Jabłonowska A., Dadlez M., Bal W., 2007. The binding constant for amyloid Aβ40 peptide interaction with human serum albumin // Biochem. Biophys. Res. Commun. V. 364. № 3. P. 714–718. https://doi.org/10.1016/j.bbrc.2007.10.080
Sadigh-Eteghad S., Sabermarouf B., Majdi A., Talebi M., Farhoudi M., Mahmoudi J., 2015. Amyloid-Beta: A crucial factor in Alzheimer’s disease // Med. Princ. Pract. V. 24. № 1. P. 1–10. https://doi.org/10.1159/000369101
Schilde L.M., Kösters S., Steinbach S., Schork K., Eisenacher M. et al., 2018. Protein variability in cerebrospinal fluid and its possible implications for neurological protein biomarker research // PLoS One. V. 13. № 11. Art. e0206478. https://doi.org/10.1371/journal.pone.0206478
Sevigny J., Chiao P., Bussière T., Weinreb P.H., Williams L. et al., 2016. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease // Nature. V. 537. № 7618. P. 50–56. https://doi.org/10.1038/nature19323
Shankar G.M., Walsh D.M., 2009. Alzheimer’s disease: Synaptic dysfunction and Abeta // Mol. Neurodegener. V. 4. № 1. Art. 48. https://doi.org/10.1186/1750-1326-4-48
Sharma K., 2019. Cholinesterase inhibitors as Alzheimer’s therapeutics (Review) // Mol. Med. Rep. V. 20. № 2. P. 1479–1487. https://doi.org/10.3892/mmr.2019.10374
Sheppard O., Coleman M., 2020. Alzheimer’s disease: Etiology, neuropathology and pathogenesis // Alzheimer’s Disease: Drug Discovery. Brisbane: Exon Publications. https://doi.org/10.36255/exonpublications.alzheimersdisease.2020.ch1
Shibata M., Yamada S., Kumar S.R., Calero M., Bading J. et al., 2000. Clearance of Alzheimer’s amyloid-β 1-40 peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier // J. Clin. Invest. V. 106. № 12. P. 1489–1499. https://doi.org/10.1172/JCI10498
Sjogren M., 2001. Both total and phosphorylated tau are increased in Alzheimer’s disease // J. Neurol. Neurosurg. Psychiatry. V. 70. № 5. P. 624–630. https://doi.org/10.1136/jnnp.70.5.624
Spires-Jones T.L., Hyman B.T., 2014. The intersection of amyloid beta and tau at synapses in Alzheimer’s disease // Neuron. V. 82. № 4. P. 756–771. https://doi.org/10.1016/j.neuron.2014.05.004
Stanyon H.F., Viles J.H., 2012. Human serum albumin can regulate amyloid-β peptide fiber growth in the brain interstitium: Implications for Alzheimer disease // J. Biol. Chem. V. 287. № 33. P. 28163–28168. https://doi.org/10.1074/jbc.C112.360800
Summers W.K., Viesselman J.O., Marsh G.M., Candelora K., 1981. Use of THA in treatment of Alzheimer-like dementia: Pilot study in twelve patients // Biol. Psychiatry. V. 16. № 2. P. 145–153. http://www.ncbi.nlm.nih.gov/pubmed/7225483
Summers W.K., Majovski L.V., Marsh G.M., Tachiki K., Kling A., 1986. Oral tetrahydroaminoacridine in long-term treatment of senile dementia, Alzheimer type // N. Engl. J. Med. V. 315. № 20. P. 1241–1245. https://doi.org/10.1056/NEJM198611133152001
Suvorina M.Y., Selivanova O.M., Grigorashvili E.I., Nikulin A.D., Marchenkov V.V. et al., 2015. Studies of polymorphism of amyloid-β42 peptide from different suppliers // J. Alzheimers Dis. V. 47. № 3. P. 583–593. https://doi.org/10.3233/JAD-150147
Tampi R.R., Forester B.P., Agronin M., 2021. Aducanumab: Evidence from clinical trial data and controversies // Drugs Context. V. 10. P. 1–9. https://doi.org/10.7573/dic.2021-7-3
Tiraboschi P., Sabbagh M.N., Hansen L.A., Salmon D.P., Merdes A. et al., 2004. Alzheimer disease without neocortical neurofibrillary tangles // Neurology. V. 62. № 7. P. 1141–1147. https://doi.org/10.1212/01.WNL.0000118212.41542.E7
Tschanz J.T., Norton M.C., Zandi P.P., Lyketsos C.G., 2013. The Cache County Study on Memory in Aging: Factors affecting risk of Alzheimer’s disease and its progression after onset // Int. Rev. Psychiatry. V. 25. № 6. P. 673–685. https://doi.org/10.3109/09540261.2013.849663
Vandesquille M., Po C., Santin M., Herbert K., Comoy E., Dhenain M., 2014. Amyloid plaques detection by MRI: Comparison of five mouse models of amyloidosis // Alzheimers Dement. V. 10. Art. 15. https://doi.org/10.1016/j.jalz.2014.05.020
Vlad S.C., Miller D.R., Kowall N.W., Felson D.T., 2008. Protective effects of NSAIDs on the development of Alzheimer disease // Neurology. V. 70. № 19. P. 1672–1677. https://doi.org/10.1212/01.wnl.0000311269.57716.63
Vusse G.J., van der, 2009. Albumin as fatty acid transporter // Drug Metab. Pharmacokinet. V. 24. № 4. P. 300–307. https://doi.org/10.2133/dmpk.24.300
Wang C., Cheng F., Xu L., Jia L., 2016. HSA targets multiple Aβ42 species and inhibits the seeding-mediated aggregation and cytotoxicity of Aβ42 aggregates // RSC Adv. V. 6. № 75. P. 71165–71175. https://doi.org/10.1039/C6RA14590F
Wang D.-S., Dickson D.W., Malter J.S., 2006. β-Amyloid degradation and Alzheimer’s disease // J. Biomed. Biotechnol. V. 2006. № 3. Art. 58406. https://doi.org/10.1155/JBB/2006/58406
Wang J., Tan L., Wang H.-F., Tan C.-C., Meng X.-F. et al., 2015. Anti-inflammatory drugs and risk of Alzheimer’s disease: An updated systematic review and meta-analysis // J. Alzheimers Dis. V. 44. № 2. P. 385–396. https://doi.org/10.3233/JAD-141506
Wang W., Dong X., Sun Y., 2019. Modification of serum albumin by high conversion of carboxyl to amino groups creates a potent inhibitor of amyloid β-protein fibrillogenesis // Bioconjug. Chem. V. 30. № 5. P. 1477–1488. https://doi.org/10.1021/acs.bioconjchem.9b00209
Whiley L., Chappell K.E., D’Hondt E., Lewis M.R., Jiménez B. et al., 2021. Metabolic phenotyping reveals a reduction in the bioavailability of serotonin and kynurenine pathway metabolites in both the urine and serum of individuals living with Alzheimer’s disease // Alzheimers Res. Ther. V. 13. № 1. Art. 20. https://doi.org/10.1186/s13195-020-00741-z
Xie B., Li X., Dong X.-Y., Sun Y., 2014. Insight into the inhibition effect of acidulated serum albumin on amyloid β‑protein fibrillogenesis and cytotoxicity // Langmuir. V. 30. № 32. P. 9789–9796. https://doi.org/10.1021/la5025197
Xie H., Guo C., 2020. Albumin alters the conformational ensemble of amyloid-β by promiscuous interactions: Implications for amyloid inhibition // Front. Mol. Biosci. V. 7. Art. 629520. https://doi.org/10.3389/fmolb.2020.629520
Yan Q., Zhang J., Liu H., Babu-Khan S., Vassar R. et al., 2003. Anti-inflammatory drug therapy alters beta-amyloid processing and deposition in an animal model of Alzheimer’s disease // J. Neurosci. V. 23. № 20. P. 7504–7509. https://doi.org/10.1523/JNEUROSCI.23-20-07504.2003
Zhang H., Liu D., Huang H., Zhao Y., Zhou H., 2018. Characteristics of insulin-degrading enzyme in Alzheimer’s disease: A meta-analysis // Curr. Alzheimer Res. V. 15. № 7. P. 610–617. https://doi.org/10.2174/1567205015666180119105446
Zhang S., Iwata K., Lachenmann M.J., Peng J.W., Li S. et al., 2000. The Alzheimer’s peptide Aβ adopts a collapsed coil structure in water // J. Struct. Biol. V. 130. № 2–3. P. 130–141. https://doi.org/10.1006/jsbi.2000.4288
Zhang W., Xiong H., Callaghan D., Liu H., Jones A. et al., 2013. Blood-brain barrier transport of amyloid beta peptides in efflux pump knock-out animals evaluated by in vivo optical imaging // Fluids Barriers CNS. V. 10. № 1. Art. 13. https://doi.org/10.1186/2045-8118-10-13
Zhao M., Guo C., 2021. Multipronged regulatory functions of serum albumin in early stages of amyloid-β aggregation // ACS Chem. Neurosci. V. 12. № 13. P. 2409–2420. https://doi.org/10.1021/acschemneuro.1c00150
Zhao Y., Marcel Y.L., 1996. Serum albumin is a significant intermediate in cholesterol transfer between cells and lipoproteins // Biochemistry. V. 35. № 22. P. 7174–7180. https://doi.org/10.1021/bi952242v
Дополнительные материалы отсутствуют.
Инструменты
Журнал общей биологии