

Журнал неорганической химии, 2022, T. 67, № 6, стр. 868-876
Термическая модификация пористых оксидных пленок, полученных анодированием алюминиево-магниевого сплава
Н. А. Ширин a, И. В. Росляков a, b, М. В. Берекчиян a, Т. Б. Шаталова a, А. В. Лукашин a, b, К. С. Напольский a, c, *
a Московский государственный университет им. М.В. Ломоносова
119991 Москва, Ленинские горы, 1, Россия
b Институт общей и неорганической химии им. Н.С. Курнакова РАН
119991 Москва, Ленинский пр-т, 31, Россия
c Московский физико-технический институт
141701 Московская обл., Долгопрудный, Институтский пер., 9, Россия
* E-mail: kirill@inorg.chem.msu.ru
Поступила в редакцию 08.02.2022
После доработки 22.02.2022
Принята к публикации 22.02.2022
- EDN: GSPENM
- DOI: 10.31857/S0044457X22060277
Аннотация
Анодное окисление алюминия в кислотных электролитах приводит к формированию на поверхности металла пористых оксидных пленок. Одним из способов управления функциональными свойствами данного материала является термическая обработка. В данной работе проведено анодирование алюминиевого сплава A5005 в 0.3 М растворе серной кислоты в кинетическом режиме. Предложена многоступенчатая программа термической обработки, которая позволяет проводить контролируемую двухстадийную кристаллизацию исходно аморфного анодного оксида алюминия с сохранением пористой структуры. На первой стадии происходит кристаллизация анодного оксида алюминия в смесь низкотемпературных полиморфных модификаций Al2O3, сопровождающаяся удалением из его структуры примесей электролита и увеличением удельной площади поверхности до 42 м2/г за счет формирования мезопористой структуры. Последующая термическая обработка при 1200°C приводит к формированию пленок α-Al2O3 со средним размером зерен 4 мкм с сохранением пористой структуры со средним диаметром пор 26 нм. Следствием кристаллизации исходно аморфного анодного оксида алюминия является увеличение его химической устойчивости на несколько порядков, что обусловливает перспективы применения разработанных методик для создания мембран, способных к функционированию в агрессивных средах, и носителей катализаторов.
ВВЕДЕНИЕ
Анодный оксид алюминия (АОА), получаемый путем электрохимического окисления (анодирования) алюминия и его сплавов в кислотных электролитах – широко известный пористый материал с гексагональной упаковкой цилиндрических каналов, расположенных перпендикулярно поверхности металла. Возможность регулировки параметров пористой структуры (диаметра пор, расстояния между порами и толщины оксидного слоя) в широком диапазоне за счет изменения условий анодирования определяет разнообразное применение AOА в науке и технике. В частности, на основе АОА созданы высокопроницаемые и высокоселективные мембраны для разделения газов [1, 2], шаблоны для нанонитей и нанотрубок различного состава [3–5], а также наноструктурированные поверхности [6, 7], каталитически [8, 9] и оптически активные материалы [10, 11], фотонные кристаллы [12].
Несмотря на широкое распространение анодирования для защиты от коррозии и декорирования изделий из алюминиевых сплавов, в большинстве научных работ, посвященных синтезу и модификации пленок АОА, в качестве исходного металла использовали высокочистый алюминий (99.99–99.999%) в виде поликристаллических фольг [13] и монокристаллов [14]. Для расширения перспектив практического применения АОА ведутся исследования особенностей формирования пористых пленок на алюминии технической чистоты. Показано, что примеси различных элементов (Cr, Cu, Fe, Mg, Mn, Si, Ti, Zn и др.) влияют на параметры структуры АОА и, в частности, приводят к меньшей упорядоченности системы пор [15, 16]. Причиной подобного поведения является отличное от высокочистого алюминия электрохимическое поведение интерметаллических частиц, которые содержатся в сплавах [17]. Кроме того, инородные включения могут приводить к неоднородной толщине оксидного слоя [18].
Термическое поведение оксидных пленок на поверхности алюминиевых сплавов менее изучено. Сплавы с высоким содержанием второго компонента (~60% титана или железа) при анодировании дают пористые пленки, состоящие из смеси аморфных оксидов. Отжиг при ~800°C приводит к формированию соответствующих кристаллических оксидов, а также смешанных оксидов (FeAl2O4, Al2TiO5) [19, 20]. Для сплавов с содержанием примесных элементов <5% рассмотрено поведение АОА при отжиге без отделения от металла в пределах 600°C [21, 22]. Авторы показали постепенное увеличение химической устойчивости пористого материала с увеличением температуры отжига. При этом кристаллизация аморфного АОА после отделения от подложки наблюдается в температурном интервале, аналогичном использованию высокочистого алюминия [21]. Таким образом, информация о термическом поведении АОА, полученного анодированием сплавов алюминия с содержанием основного компонента >95%, носит фрагментарный характер, а данные о кристаллизации в фазу α-Al2O3 отсутствуют.
Цель работы – разработка методики управляемой кристаллизации АОА, полученного анодированием алюминиевого сплава A5005 в сернокислом электролите, в фазу α-Al2O3 с сохранением электрохимически сформированной системы пор. Прослежены изменения морфологии, фазового состава и кристаллической структуры АОА в диапазоне температур до 1200°C. Впервые на различных стадиях отжига исследованы практически значимые функциональные характеристики (удельная площадь поверхности, химическая устойчивость) пористых пленок АОА, полученных анодированием алюминия технической чистоты.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Исходным материалом для получения пористых пленок АОА служил сплав А5005 (соответствует марке АМг1 по ГОСТ 4784-97) в виде листов с толщиной 2 мм. Данный сплав содержит от 97 до 97.6 мас. % алюминия и в качестве основных легирующих компонентов – от 0.5 до 1.1 мас. % магния, не более 0.7 мас. % железа и не более 0.3 мас. % кремния. На предварительном этапе для очистки поверхности металла от механических и химических загрязнений, а также уменьшения шероховатости проводили электрохимическую полировку по ранее предложенной методике [23].
Анодное окисление металла осуществляли в тефлоновой двухэлектродной электрохимической ячейке при 25 В с использованием 0.3 М раствора H2SO4 в качестве электролита, который охлаждали до ~1°C и интенсивно перемешивали. Лист сплава А5005, который выступал в качестве анода, прижимали ко дну ячейки с помощью винтового механизма с уплотнением в виде витонового кольца (внутренний диаметр 3.2 см). Таким образом, электролит контактировал лишь с одной поверхностью листа алюминиевого сплава. Катодом служило титановое кольцо диаметром 3 см, расположенное на расстоянии 8 см от анода. Продолжительность процесса контролировали кулонометрически, используя экспериментально определенный удельный заряд, равный 2.2 Кл/(см2 мкм). Анодирование прекращали, когда толщина АОА достигала 50 мкм.
После анодирования оставшийся алюминий удаляли в растворе, содержащем 10 об. % Br2 в CH3OH, при комнатной температуре. Для получения пленок со сквозными открытыми порами проводили химическое травление сплошного барьерного слоя на нижней поверхности АОА в 5 мас. % H3PO4 при температуре 60°C в течение 100 с.
Отжиг АОА проводили в печи Nabertherm L5/12 на воздухе между двумя корундовыми пластинами для предотвращения механических деформаций (изгибания). Температурный режим отжига подробно рассмотрен в разделе “Обсуждение результатов”.
Синхронный термический анализ (СТА) с масс-спектрометрией отходящих газов проводили при скорости нагрева 5 град/мин на термоанализаторе Netzsch STA 409 PC Luxx, совмещенном с квадрупольным масс-спектрометром Netzch QMS 403C Aëolos. Измерения осуществляли в динамической воздушной атмосфере при скорости потока 30 мл/мин. Масса навески составляла около 40 мг. Температуры начала фазовых переходов установлены из данных дифференциальной сканирующей калориметрии (ДСК) по пересечению касательных к базовой линии и к начальной ветви кривой термического эффекта [24]. Погрешность измерения температуры при ДСК составляет ~1°С. Для каждого из исследованных образцов анализ проводили однократно.
Рентгенофазовый анализ (РФА) проводили на дифрактометре Rigaku D/Max-2500, используя CuKα-излучение с длиной волны 1.5418 Å, в диапазоне 2θ от 10° до 90° c шагом сканирования 0.02°. Идентификацию фаз осуществляли с использованием базы данных ICDD PDF2.
Удельную площадь поверхности и распределение пор по размерам измеряли методом капиллярной конденсации азота при 77 K с использованием анализатора поверхности Quanta Chrome NOVA 4200E. Предварительно АОА (~200 мг) вакуумировали при температуре 300°C и остаточном давлении ~1 мбар в течение 3 ч. В случае анализа неотожженного АОА стадию дегазации проводили в течение 10 ч при 100°C. Расчеты целевых характеристик проводили по методу Баррета–Джойнера–Халенды (Barrett–Joyner–Halenda – BJH) [25]. Перед проведением СТА, РФА и капиллярной конденсации азота пленки АОА перетирали в порошок в агатовой ступке.
Растровую электронную микроскопию (РЭМ) проводили на микроскопе Carl Zeiss NVision 40. Предварительно на поверхность оксидных пленок наносили слой хрома толщиной 5 нм с помощью установки магнетронного напыления Quorum Technologies Q150T. РЭМ изображения для вычисления геометрических параметров пористой структуры обрабатывали в программах ImageJ [26] и Statistics2D [27].
Устойчивость АОА в агрессивных средах оценивали путем расчета степени растворения АОА (x, мас. %) в процессе его выдерживания в 1 М растворе HCl при комнатной температуре (~26°C). Величину х определяли как отношение массы растворенного АОА к исходной массе навески. Для этого измеряли временную зависимость концентрации ионов Al3+ в растворе с помощью масс-спектрометра Perkin Elmer Elan DRC II с ионизацией пробы индуктивно-связанной аргоновой плазмой. Используя полученные значения концентрации алюминия (СAl, мг/л), массу навески АОА (mАОА, г) и найденную при термогравиметрическом анализе массовую долю оксида алюминия в пленках АОА (${{{{\omega }}}_{{{\text{A}}{{{\text{l}}}_{2}}{{{\text{O}}}_{3}}}}},$ мас. %), рассчитывали величину x по формуле:
где Vисх – исходный объем раствора, мл; Vi – объем i-ой аликвоты, мл; M – молярные массы веществ, г/моль.ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Данные СТА демонстрируют термическое превращение в три стадии, которые являются типичными для АОА, полученного анодированием высокочистого алюминия [28, 29]. Потеря массы в ~2% на первой стадии (ниже 800°C) соответствует удалению сорбированной и/или химически связанной воды (рис. 1a). Вторая стадия включает резкую потерю массы в диапазоне от 910 до 960°C, которая является следствием разложения сульфат-ионов, внедренных в структуру АОА из электролита. В пользу этого свидетельствует максимум ионного тока для массового числа 64, которое соответствует выделению SO2 (рис. 1б). Экзотермический максимум c температурой начала соответствующего процесса 926°C возникает вследствие кристаллизации исходно аморфного АОА в смесь низкотемпературных полиморфных модификаций Al2O3. Согласно данным РФА (рис. 2), после отжига при 926°C АОА в основном состоит из γ-Al2O3. Однако количественный фазовый анализ АОА, подвергнутого отжигу в данном диапазоне температур, затруднен вследствие близкой кристаллической структуры низкотемпературных полиморфных модификаций Al2O3 с многочисленными дефектами упаковки [30]. Указанная температура кристаллизации близка к температуре данного фазового перехода для АОА, полученного анодированием высокочистого алюминия в электролитах на основе серной кислоты в аналогичных условиях [31, 32]. Напротив, анодирование алюминия технической чистоты (сплав Al6082) в щавелевой кислоте приводит к понижению температуры кристаллизации АОА до 882°C [21]. Снижение температуры кристаллизации АОА при переходе от сернокислого к щавелевокислому электролиту типично для оксидных пленок, полученных на поверхности высокочистого алюминия [29, 33, 34].
Рис. 1.
Результаты исследования АОА с помощью термогравиметрического анализа (сплошная линия), дифференциальной сканирующей калориметрии (пунктирная линия) (а) и масс-спектрометрии отходящих газов для массового числа 64 (б).
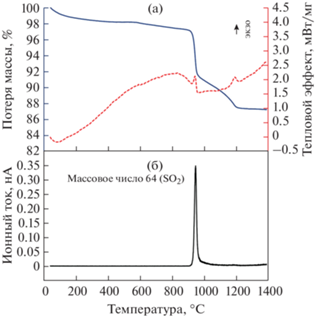
Рис. 2.
Дифрактограммы АОА до отжига (1) и после отжига при 926°C в течение 30 мин (2) и 1200°C в течение 1 ч (3).
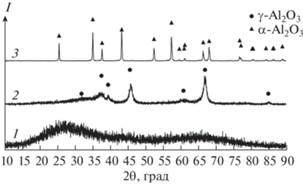
Последующий нагрев сопровождается плавной потерей массы вплоть до температуры 1200°C. Общее количество серосодержащих примесей, рассчитанное из потери массы в интервале температур от 800 до 1200°C, составляет 9.9 мас. %. Данное значение согласуется с массовой долей серосодержащих примесей в АОА, полученном анодированием высокочистого алюминия в аналогичных условиях [29, 32]. Последующая кристаллизация проявляется в виде экзотермического максимума на кривой ДСК с температурой начала процесса 1153°C. По данным РФА (рис. 2), указанный максимум соответствует превращению АОА в фазу α-Al2O3.
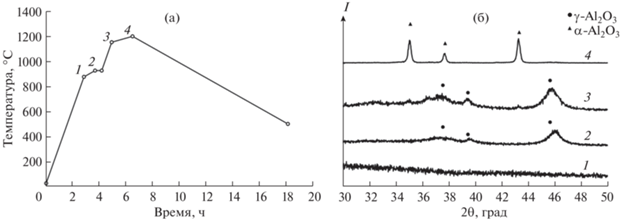
Для кристаллизации АОА в фазу α-Al2O3 с сохранением пористой структуры была разработана семиступенчатая программа термической обработки (рис. 3а) с использованием подходов, аналогичных описанным в работах [32, 35]. В основе программы лежат температуры фазовых переходов, определенные по результатам ДТА. Кристаллизация аморфного АОА и переход низкотемпературных полиморфных модификаций Al2O3 в фазу α-Al2O3 происходят на стадиях с малой скоростью нагрева, что подтверждается данными РФА (рис. 3б). В частности, нагрев до температуры 876°C не приводит к появлению дифракционных максимумов. Напротив, после медленного нагрева от 876 до 926°C со скоростью 1 град/мин отчетливо видна кристаллизация АОА. Последующий нагрев до 1153°C соответствует непрерывному изменению соотношения низкотемпературных полиморфных модификаций Al2O3 и/или совершенствованию их структуры, о чем свидетельствует увеличение интенсивности и изменение формы наблюдаемых дифракционных максимумов. Медленный нагрев до 1200°C со скоростью 0.5 град/мин приводит к появлению интенсивных дифракционных максимумов, соответствующих фазе α-Al2O3.
Рис. 3.
Разработанная программа термической обработки, приводящая к кристаллизации АОА в фазу α-Al2O3 с сохранением исходной пористой структуры (а). Дифрактограммы, иллюстрирующие фазовый состав на различных стадиях термической обработки: 876 (1), 926 (2), 1153 (3) и 1200°C (4) (б). Нумерация кривых на панели (б) соответствует обозначениям на панели (а).
На рис. 4 представлены фотографии пористой пленки АОА после удаления алюминиевой основы, а также после кристаллизации при температурах 926 и 1200°C. При термической обработке сохраняется целостность оксидной пленки, растрескивания, скручивания и других механических деформаций не происходит. Наблюдается постепенная потеря прозрачности пленок, что характерно для АОА при кристаллизации и переходе в фазу α-Al2O3 [35].
Рис. 4.
Фотографии пленок АОА после удаления алюминиевой основы (а) и последующего отжига при 926 (б) и 1200°С (в).

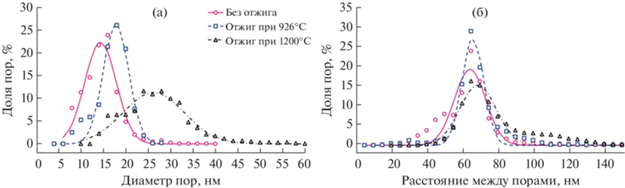
АОА исследовали с помощью РЭМ (рис. 5) с последующим статистическим анализом полученных изображений (рис. 6). В исходной пористой структуре АОА цилиндрические каналы на нижней поверхности имеют средний диаметр 14 ± 4 нм, а среднее расстояние между ними (Dint) составляет 64 ± 10 нм. Кристаллизация при 926°C приводит к увеличению диаметра пор примерно на 25% до 18 ± 3 нм (рис. 6а). При последующей кристаллизации в фазу α-Al2O3 диаметр каналов увеличивается еще сильнее и составляет 26 ± 8 нм (увеличение примерно на 85% относительно исходного значения). В то же время при указанных фазовых переходах расстояние между порами остается без изменений в пределах погрешности (рис. 6б).
Рис. 5.
РЭМ изображения верхней (а, в, д) и нижней (б, г, е, ж) поверхности АОА после удаления барьерного слоя и последующего отжига в различных условиях. Представлены изображения исходного АОА (а, б), после кристаллизации в смесь низкотемпературных полиморфных модификаций Al2O3 (в, г) и в фазу α-Al2O3 (д, е, ж).

Рис. 6.
Результаты статистического анализа РЭМ изображений АОА на различных стадиях термической обработки: распределение диаметра пор (а) и расстояния между порами (б). Приведены данные для нижней поверхности пористых пленок после удаления барьерного слоя. Линии соответствуют аппроксимации экспериментальных данных распределением Гаусса.
На РЭМ изображении нижней поверхности АОА при малом увеличении (рис. 5ж) виден набор мозаичных зерен со средним размером (эквивалентным диаметром) 4 ± 1 мкм. Каждое из наблюдаемых зерен пронизано вертикальными цилиндрическими каналами. Ранее аналогичная морфология была визуализирована с помощью РЭМ и дифракции обратно рассеянных электронов [29], а также флуоресцентной микроскопии [36] для АОА, полученного анодированием высокочистого алюминия в аналогичных условиях с последующей термической обработкой для кристаллизации в фазу α-Al2O3.
В настоящей работе исследована термическая устойчивость пористого α-Al2O3 при высоких температурах. На рис. 7 представлена серия РЭМ изображений, полученных для нижней поверхности АОА после удаления барьерного слоя и кристаллизации в фазу α-Al2O3 с различным временем выдержки при температуре 1200°C. Для количественного описания пористой структуры использован параметр p, который характеризует количество каналов на единицу площади. Отметим, что для гексагональной упаковки с Dint = 64 нм p = = 282 мкм−2. Уже после выдержки в течение 18 мин начинается спекание материала с исчезновением части каналов, однако величина p уменьшается незначительно и составляет ~260 мкм−2. Получасовая выдержка при 1200°C приводит к двукратному уменьшению p до ~130 мкм−2. После часовой выдержки пористая структура значительно деградирует (p ~ 100 мкм−2). Данные результаты следует иметь в виду при практическом использовании АОА в фазе α-Al2O3. Отметим, что наблюдаемая термическая устойчивость АОА, полученного анодированием алюминия технической чистоты в 0.3 М серной кислоте при 25 В, оказывается значительно ниже, чем для крупнопористого АОА [35]. Это может быть связано с малым диаметром каналов и, как следствие, их быстрой коалесценцией уже при 1200°C.
Рис. 7.
РЭМ изображения нижней поверхности АОА после удаления барьерного слоя и последующей кристаллизации в фазу α-Al2O3 и выдержкой при 1200°C в течение 2 (а), 18 (б), 30 (в) и 60 мин (г).

В качестве альтернативного подхода к аттестации пористой структуры АОА использован метод капиллярной конденсации азота при 77 K. Результаты обработки изотерм по методу BJH (рис. 8а) показывают наличие каналов диаметром ~14 нм для исходного АОА, что хорошо согласуется с данными РЭМ (рис. 6а). Второй максимум при ~4 нм может соответствовать дефектам, формирующимся при окислении интерметаллических частиц. Удельная площадь поверхности (S) при этом составляет около 13 м2/г. Отжиг при 926°C в течение 30 мин приводит к увеличению S до 42 м2/г за счет образования в стенках каналов мезопор со средним диаметром ~5 нм. Согласно [37], причиной формирования мезопор на данной стадии термической обработки является удаление примесей электролита путем их термического разложения с образованием летучих соединений. Аналогичное увеличение S после первой стадии кристаллизации характерно для АОА, полученного анодированием высокочистого алюминия в электролитах на основе серной и щавелевой кислот [32, 33, 37, 38]. Отметим, что дефекты анодирования интерметаллических частиц (максимум при ~4 нм), а также электрохимически сформированные каналы (максимум при ~16 нм) не претерпевают существенных изменений в процессе отжига. При этом диаметр электрохимически сформированных каналов совпадает с диаметром каналов АОА, который получен анодированием высокочистого алюминия в аналогичных условиях [32].
Рис. 8.
Распределение пор по размерам согласно данным капиллярной конденсации азота при 77 K на различных стадиях термической обработки АОА: (а) до и после кристаллизации в смесь низкотемпературных полиморфных модификаций Al2O3 и (б) после кристаллизации в фазу α-Al2O3 (время выдержки при 1200°C составляло 2 мин). S – удельная площадь поверхности, м2/г; D – диаметр пор, нм.
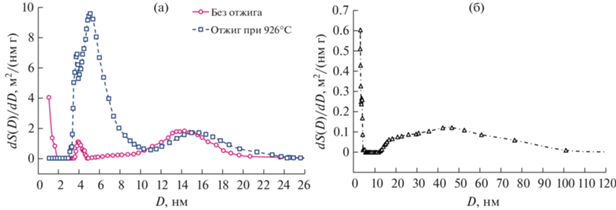
Отжиг при 1200°C приводит к коалесценции мезопор, что выражается в уменьшении удельной площади поверхности до 6 м2/г. Данный эффект наблюдали и в случае получения АОА анодированием высокочистого алюминия в аналогичных условиях [32]. Также можно видеть частичное срастание электрохимически сформированных каналов между собой, что заметно по уширению распределения пор по диаметру и сдвигу максимума распределения в сторону бо́льших значений из-за разрушения стенок между порами. Указанные структурные изменения при кристаллизации в фазу α-Al2O3 хорошо заметны на РЭМ изображениях (рис. 7). Отметим, что различия в распределениях пор по размерам на данной стадии по результатам капиллярной конденсации азота (рис. 8б) и РЭМ (рис. 6а) могут свидетельствовать о некотором отличии пористой структуры на поверхности и в толще мембраны.
Аттестацию химической устойчивости АОА после различных стадий термической обработки проводили путем травления пленок в 1 М HCl при комнатной температуре (~26°C) и последующего анализа массы растворенного Al2O3. Рис. 9 иллюстрирует временные зависимости степени растворения АОА. Для исходного материала наблюдается постоянная скорость растворения (v), которая составляет 23.2(9) мас. %/ч (рис. 9а). То есть в указанных условиях аморфный АОА полностью растворяется менее чем за 5 ч. Наблюдаемая высокая скорость растворения аморфного АОА согласуется с ранее полученными данными в кислых и щелочных средах [39–41]. Термическая обработка АОА значительно увеличивает его химическую стабильность [39, 41, 42]. В частности, после кристаллизации в смесь низкотемпературных полиморфных модификаций Al2O3 степень растворения составляет лишь 3 мас. % после 7 сут нахождения в 1 М растворе HCl (рис. 9б). Перевод АОА в фазу α-Al2O3 уменьшает общую степень растворения материала на порядок до 0.2 мас. % при двукратном увеличении времени травления (рис. 9в).
Рис. 9.
Зависимость степени растворения АОА от времени выдержки в 1 М растворе соляной кислоты при комнатной температуре (~26°C) для исходного АОА (а), после кристаллизации в смесь низкотемпературных полиморфных модификаций Al2O3 (отжиг при 926°C) (б) и в фазу α-Al2O3 (отжиг при 1200°C) (в). Пунктирные линии соответствуют линейной аппроксимации экспериментальных данных.
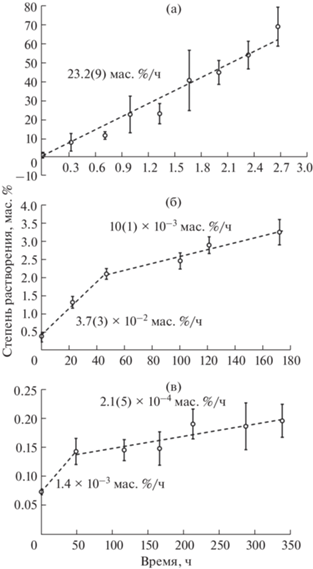
На зависимостях степени растворения от времени выдержки после кристаллизации АОА можно выделить два линейных участка. Большую скорость растворения на начальном этапе (v = 3.7(3) × × 10–2 мас. %/ч) в случае АОА, отожженного при 926°C в течение 30 мин (рис. 9б), можно связать с меньшей химической стойкостью одной из примесных низкотемпературных полиморфных модификаций Al2O3 по сравнению с основной фазой γ-Al2O3 (v = = 10(1) × 10–3 мас. %/ч). Аналогично, остаточные примеси низкотемпературных полиморфных модификаций Al2O3 обусловливают большую величину v ~ 1 × 10–3 мас. %/ч на начальной стадии химического травления пористого α-Al2O3 (рис. 9в), который при больших временах выдержки проявляет исключительно высокую химическую стабильность (v = 2.1(5) × 10–4 мас. %/ч).
ЗАКЛЮЧЕНИЕ
Анодированием алюминиевого сплава A5005 в 0.3 М серной кислоте при напряжении 25 В получены пористые пленки АОА со средним диаметром каналов 14 нм и средним расстоянием между их центрами 64 нм. Исследование термического поведения показало кристаллизацию исходно аморфного АОА в смесь низкотемпературных полиморфных модификаций Al2O3 при 926°C, которая сопровождается удалением из структуры примесей электролита и увеличением удельной площади поверхности до 42 м2/г за счет образования в стенках каналов мезопор. Использование разработанной программы отжига позволяет сохранить исходную пористую структуру при последующей кристаллизации в α-Al2O3 при 1153°C. АОА в фазе α-Al2O3 характеризуется крупнокристаллической структурой со средним размером зерен ~4 мкм, которые пронизаны параллельными цилиндрическими каналами с диаметром ~26 нм.
Термическая обработка АОА приводит к снижению скорости его растворения в 1 М HCl при переходе в смесь низкотемпературных полиморфных модификаций Al2O3 более чем на три порядка, а при кристаллизации в α-Al2O3 еще на порядок. Показанная химическая стойкость АОА в сочетании с уникальной пористой структурой и, в случае кристаллизации при 926°C, с высокой удельной площадью поверхности открывают перспективы использования АОА с различным фазовым составом для создания мембран и носителей катализаторов. Предложенные в работе подходы к получению химически стойких мембран (анодирование алюминия технической чистоты в сернокислом электролите в кинетическом режиме) обусловливают простоту масштабирования проведенных экспериментов.
Список литературы
Chernova E.A., Petukhov D.I., Kapitanova O.O. et al. // Nanosyst. Phys. Chem. Math. 2018. V. 9. № 5. P. 614. https://doi.org/10.17586/2220-8054-2018-9-5-614-621
Roslyakov I. V., Petukhov D.I., Napolskii K.S. // Nanotechnology. 2021. V. 32. № 33. P. 33LT01. https://doi.org/10.1088/1361-6528/ABFEEA
Мистонов А.А., Дубицкий И.С., Елмекави А.Х.А. и др. // Физ. тверд. тела 2021. Т. 63. № 7. С. 881.
Ryzhkov I.I., Kharchenko I.A., Mikhlina E. V. et al. // Int. J. Heat Mass Transf. 2021. V. 176. P. 121414. https://doi.org/10.1016/j.ijheatmasstransfer.2021.121414
Давыдов А.Д., Волгин В.М. // Электрохимия. 2016. Т. 52. № 9. С. 905. [Davydov A.D., Volgin V.M. // Russ. J. Electrochem. 2016. V. 52. № 9. P. 806. https://doi.org/10.1134/S1023193516090020]
Валеев Р.Г., Тригуб А.Л., Бельтюков А.Н. и др. // Поверхн. рентген. синхротр. нейтр. исслед. 2019. № 2. С. 28. [Valeev R.G., Trigub A.L., Beltiukov A.N. et al. // J. Surf. Invest.: X-Ray, Synchrotron Neutron Tech. 2019. V. 13. № 1. P. 92. https://doi.org/10.1134/S1027451019010373]
Гасенкова И.В., Андрухович И.М., Ткачев В.В. // Поверхн. рентген. синхротр. нейтр. исслед. 2019. № 8. С. 35. [Gasenkova I.V., Andrukhovich I.M., Tkachev V.V. // J. Surf. Invest.: X-Ray, Synchrotron Neutron Tech. 2019. V. 13. № 4. P. 700. https://doi.org/10.1134/S1027451019040232]
Кокатев А.Н., Лукиянчук И.В., Яковлева Н.М. и др. // Физикохим. поверхн. защ. матер. 2016. Т. 52. № 5. С. 517. [Kokatev A.N., Lukiyanchuk I. V., Yakovleva N.M. et al. // Prot. Met. Phys. Chem. Surf. 2016. V. 52. № 5. P. 832. https://doi.org/10.1134/S2070205116050130]
Roslyakov I.V., Kolesnik I.V., Evdokimov P.V. et al. // Sens. Actuators B. 2021. V. 330. P. 129307. https://doi.org/10.1016/J.SNB.2020.129307
Ибраев Н.Х., Зейниденов А.К., Аймуханов А.К. и др. // Квантовая электрон. 2015. Т. 45. № 7. С. 663. [Ibrayev N.K., Zeinidenov A.K., Aimukhanov A.K. et al. // Quantum Electron. 2015. V. 45. № 7. P. 663. https://doi.org/10.1070/qe2015v045n07abeh015533]
Помозов А.Р., Колмычек И.А., Ганьшина Е.А. и др. // Физ. тверд. тела. 2018. Т. 60. № 11. С. 2224. [Pomozov A.R., Kolmychek I.A., Gan’shina E.A. et al. // Phys. Solid State 2018. V. 60. № 11. P. 2264. https://doi.org/10.1134/S1063783418110264]
Napolskii K.S., Noyan A.A., Kushnir S.E. // Opt. Mater. 2020. V. 109. P. 110317. https://doi.org/10.1016/j.optmat.2020.110317
Леонтьев А.П., Росляков И.В., Веденеев А.С. и др. // Поверхн. рентген. синхротр. нейтр. исслед. 2016. № 5. С. 88. [Leontiev A.P., Roslyakov I.V., Vedeneev A.S. et al. // J. Surf. Invest.: X-Ray, Synchrotron Neutron Tech. 2016. V. 10. № 3. P. 548. https://doi.org/10.1134/S1027451016030113]
Росляков И.В., Кошкодаев Д.С., Лебедев В.А. и др. // Поверхн. рентген. синхротр. нейтр. исслед. 2019. № 10. С. 74. [Roslyakov I.V., Koshkodaev D.S., Lebedev V.A. et al. // J. Surf. Invest.: X-Ray, Synchrotron Neutron Tech. 2019. V. 13. № 5. P. 955. https://doi.org/10.1134/S1027451019050343]
Zaraska L., Sulka G.D., Szeremeta J. et al. // Electrochim. Acta 2010. V. 55. № 14. P. 4377. https://doi.org/10.1016/j.electacta.2009.12.054
Григорьев С.В., Григорьева Н.А., Сыромятников А.В. и др. // Письма ж. эксп. теор. физ. 2007. Т. 85. № 9–10. С. 549. [Grigor’ev S.V., Grigor’eva N.A., Syromyatnikov A.V. et al. // JETP Lett. 2007. V. 85. № 9. P. 449. https://doi.org/10.1134/S0021364007090081]
Montero-Moreno J.M., Sarret M., Müller C. // Microporous Mesoporous Mater. 2010. V. 136. № 1–3. P. 68. https://doi.org/10.1016/j.micromeso.2010.07.022
Chung C.-K., Liao M.-W., Lee C.-T. et al. // Nanoscale Res. Lett. 2011. V. 6. № 1. P. 1. https://doi.org/10.1186/1556-276X-6-596
Stepniowski W.J., Choi J., Yoo H. et al. // J. Electroanal. Chem. 2016. V. 771. P. 37. https://doi.org/10.1016/j.jelechem.2016.04.010
Степанова К.В., Яковлева Н.М., Кокатев А.Н. и др. // Поверхн. Рентген. синхротр. нейтр. исслед. 2016. № 9. С. 54. [Stepanova K.V., Yakovleva N.M., Kokatev A.N. et al. // J. Surf. Invest.: X-Ray, Synchrotron Neutron Tech. 2016. V. 10. № 5. P. 933. https://doi.org/10.1134/S102745101605013X]
Kozhukhova A.E., du Preez S.P., Bessarabov D.G. // Surf. Coat. Technol. 2020. V. 383. P. 125234. https://doi.org/10.1016/j.surfcoat.2019.125234
Chen C.C., Chen J.H., Chao C.G. // Jpn. J. Appl. Phys. 2005. V. 44. № 3R. P. 1529. https://doi.org/10.1143/JJAP.44.1529
Гордеева Е.О., Росляков И.В., Садыков А.И. и др. // Электрохимия. 2018. Т. 54. № 11. С. 999. [Gordeeva E.O., Roslyakov I. V., Sadykov A.I. et al. // Russ. J. Electrochem. 2018. V. 54. № 11. P. 990. https://doi.org/10.1134/S1023193518130165]
Höhne G.W.H., Hemminger W.F., Flammersheim H.-J. Differential Scanning Calorimetry. Heidelberg: Springer, 2003. 298 p. https://doi.org/10.1007/978-3-662-06710-9
Barrett E.P., Joyner L.G., Halenda P.P. // J. Am. Chem. Soc. 2002. V. 73. № 1. P. 373. https://doi.org/10.1021/JA01145A126
Schneider C.A., Rasband W.S., Eliceiri K.W. // Nat. Methods. 2012. V. 9. № 7. P. 671. https://doi.org/10.1038/nmeth.2089
http://eng.fnm.msu.ru/software/
Cho S.Y., Kim J.W., Bu S.D. // J. Korean Phys. Soc. 2015. V. 66. № 9. P. 1394. https://doi.org/10.3938/JKPS.66.1394
Mata-Zamora M.E., Saniger J.M. // Rev. Mex. Fis. 2005. V. 51. № 5. P. 502.
Tsybulya S. V., Kryukova G.N. // Phys. Rev. B 2008. V. 77. № 2. P. 024112. https://doi.org/10.1103/PhysRevB.77.024112
Воробьева А.И., Шиманович Д.Л., Сычева О.А. // Микроэлектроника 2018. № 1. С. 45. [Vorob’eva A.I., Shimanovich D.L., Sycheva O.A. // Russ. Microelectron. 2018. V. 47. № 1. P. 40. https://doi.org/10.1134/S1063739718010080]
Roslyakov I.V., Shirin N.A., Berekchiian M.V. et al. // Microporous Mesoporous Mater. 2020. V. 294. P. 109840. https://doi.org/10.1016/j.micromeso.2019.109840
Гасенкова И.В., Остапенко Е.В. // Поверхн. pентген. синхротр. нейтр. исслед. 2013. Т. 2013. № 6. С. 39. [Gasenkova I.V., Ostapenko E.V. // J. Surf. Invest.: X-Ray, Synchrotron Neutron Tech. 2013. V. 7. № 3. P. 536. https://doi.org/10.1134/S1027451013030245]
Росляков И.В., Напольский К.С., Евдокимов П.В. и др. // Наносист.: физ. хим. мат. 2013. Т. 4. № 1. С. 65.
Masuda T., Asoh H., Haraguchi S. et al. // Materials 2015. V. 8. № 3. P. 1350. https://doi.org/10.3390/MA8031350
Chernyakova K., Karpicz R., Rutkauskas D. et al. // Phys. Status Solidi A 2018. V. 215. № 16. P. 1700892. https://doi.org/10.1002/PSSA.201700892
Roslyakov I.V., Kolesnik I.V., Levin E.E. et al. // Surf. Coat. Technol. 2020. V. 381. P. 125159. https://doi.org/10.1016/j.surfcoat.2019.125159
Mardilovich P.P., Govyadinov A.N., Mukhurov N.I. et al. // J. Membr. Sci. 1995. V. 98. № 1–2. P. 131. https://doi.org/10.1016/0376-7388(94)00184-Z
Mardilovich P.P., Govyadinoy A.N., Mazurenko N.I. et al. // J. Membr. Sci. 1995. V. 98. № 1–2. P. 143. https://doi.org/10.1016/0376-7388(94)00185-2
Садыков А.И., Леонтьев А.П., Кушнир С.Е. и др. // Журн. неорган. химии. 2021. Т. 66. № 2. С. 265. [Sadykov A.I., Leontev A.P., Kushnir S.E. et al. // Russ. J. Inorg. Chem. 2021. V. 66. № 2. P. 258. https://doi.org/10.1134/S0036023621020182]
Lee C.-W., Kang H.-S., Chang Y.-H. et al. // Korean J. Chem. Eng. 2000 17:3 2000. V. 17. № 3. P. 266. https://doi.org/10.1007/BF02699038
Santos A., Kumeria T., Wang Y. et al. // Nanoscale. 2014. V. 6. № 17. P. 9991. https://doi.org/10.1039/C4NR01422G
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии