

Журнал неорганической химии, 2022, T. 67, № 6, стр. 750-760
Формирование в условиях гидротермально-микроволнового синтеза и оптические свойства фазы пирохлора в системе Bi2O3‒Fe2O3‒WO3‒(H2O)
М. С. Ломакин a, b, *, О. В. Проскурина a, c, А. А. Левин a, А. А. Сергеев d, А. А. Леонов d, В. Н. Неведомский a, С. С. Вознесенский d
a Физико-технический институт им. А.Ф. Иоффе
194021 Санкт-Петербург, ул. Политехническая, 26, Россия
b Санкт-Петербургский государственный электротехнический университет “ЛЭТИ”
197376 Санкт-Петербург, ул. Профессора Попова, 5, Россия
c Санкт-Петербургский государственный технологический институт (технический университет)
190013 Санкт-Петербург, Московский пр-т, 26, Россия
d Институт автоматики и процессов управления ДВО РАН
690041 Владивосток, ул. Радио, 5, Россия
* E-mail: lomakinmakariy@gmail.com
Поступила в редакцию 02.11.2021
После доработки 25.11.2021
Принята к публикации 29.11.2021
- EDN: DLLIPZ
- DOI: 10.31857/S0044457X22060149
Аннотация
Методом гидротермально-микроволнового синтеза в системе Bi2O3‒Fe2O3‒WO3‒(H2O) получены и охарактеризованы композиты состава (Bi, Fe, ◻)2(Fe, W)2O6O′δ (пирохлор, далее BFWO)/аморфная фаза. Установлено, что степень превращения аморфной фазы в кристаллическую BFWO возрастает с ~0.08 до ~0.82 при увеличении продолжительности изотермической (180°С) выдержки от 1 с до 5 мин. С видом этой зависимости хорошо коррелирует характер изменения среднего размера кристаллитов фазы BFWO (оцененного графическим методом Вильямсона–Холла), который возрастает от 270(20) до ~600 нм. Показано, что во всех образцах присутствует два морфологических мотива, один из которых представлен частицами, средний размер которых коррелирует со средним размером кристаллитов фазы BFWO, а другой – агломератами размером ~100 нм, составленными из частиц аморфной фазы. Крупные частицы имеют структуру типа ядро‒оболочка, в которой ядром являются кристаллы BFWO, а оболочкой ‒ слой аморфной фазы. Ширина запрещенной зоны (Eg) полученных композиционных материалов для прямых разрешенных электронных переходов находится в диапазоне 2.26‒2.40 эВ и уменьшается с увеличением доли кристаллической фазы BFWO в композите.
ВВЕДЕНИЕ
Последние десятилетия объектами повышенного интереса исследователей являются сложнооксидные материалы со структурой кубического пирохлора [1‒4]. Тройные пирохлоры, содержащие висмут и d-элементы, обладают широким спектром электрофизических [5‒7], магнитных [8‒10], каталитических [11‒13] и оптических [14, 15] свойств, что определяет перспективы их применения, например, в устройствах электронной техники [16, 17], фотовольтаике и фотокатализе [18, 19]. Комплекс функциональных свойств, проявляемых материалами на основе фазы пирохлора, будет в значительной степени определяться фазовым и химическим составом, а также морфологическими и размерными параметрами частиц и кристаллитов. С кристаллохимической точки зрения структура пирохлора состоит из двух взаимопроникающих подрешеток (A2O' и B2O6), качественный и количественной состав которых можно варьировать в широких пределах, не выходя из области гомогенности этой фазы [20]. В связи с этим активно ведутся работы по синтезу новых тройных висмутсодержащих пирохлоров [21‒23], в том числе исследуются пределы области гомогенности уже известных соединений с этой структурой [24‒26]. В большинстве работ фазу со структурой пирохлора получают методом твердофазного синтеза [5, 7‒9, 20‒22, 24‒26], и только некоторые работы посвящены синтезу и изучению формирования этой фазы в условиях гидротермальной обработки [12, 15, 27‒29]. Поэтому тройная оксидная система Bi2O3‒Fe2O3‒WO3‒(H2O) плохо изучена, и лишь отдельные работы посвящены синтезу, исследованию структуры и свойств кубической фазы пирохлора, формирующейся в этой системе [15, 30, 31].
Целью данной работы является изучение процесса формирования композитов на основе фазы BFWO в условиях гидротермально-микроволновой обработки и определение их оптических свойств в зависимости от соотношения сосуществующих фаз (BFWO/аморфная фаза).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
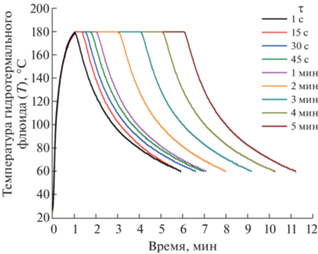
Синтез образцов проводили в соответствии со следующей методикой. Для получения одного образца 3 ммоль кристаллогидрата нитрата висмута(III) Bi(NO3)3 ⋅ 5H2O (ос. ч.) и 2.19 ммоль кристаллогидрата нитрата железа(III) Fe(NO3)3 ⋅ ⋅ 9H2O (ч.) было растворено в 7.5 мл 6 M HNO3 (ос. ч.), после чего в полученный раствор было добавлено 37.5 мл дистиллированной воды. Далее 6 ммоль кристаллогидрата вольфрамата(VI) натрия Na2WO4 ⋅ 2H2O (ос. ч.) растворяли в 38 мл дистиллированной воды и полученный раствор по каплям добавляли в перемешиваемый кислый раствор нитратов висмута и железа, туда же добавляли ~20 мл дистиллированной воды, которой был промыт стакан, в котором находился раствор вольфрамата натрия. Через 1 ч непрерывного перемешивания образовавшейся суспензии к ней по каплям добавляли раствор 4 M NaOH (~10–15 мл) до pH ~ 4. Полученную таким образом суспензию прекурсора (~110–120 мл) после дополнительного перемешивания в течение ~1 ч переносили в автоклав объемом 30 мл, представляющий собой стеклянную ампулу с герметичной крышкой (степень заполнения объема ~25% при загрузке 8 мл суспензии прекурсора), который затем помещали в установку для микроволнового нагрева Monowave 400 (Anton Paar). После этапа гидротермально-микроволновой обработки, программа которой приведена ниже, суспензию извлекали из автоклава, потом его промывали и подвергали следующие 8 мл суспензии прекурсора гидротермально-микроволновой обработке при тех же параметрах (эту процедуру повторяли до тех пор, пока весь объём суспензии прекурсора, продолжающейся перемешиваться, не был использован для получения одного образца). Для получения следующего образца всю описанную выше методику синтеза повторяли заново. Нагрев проводили со скоростью 180 град/мин до температуры 180°С. Продолжительность изотермической выдержки при 180°С варьировали следующим образом: 1, 15, 30, 45 с и 1, 2, 3, 4, 5 мин. Контроль температуры осуществляли по показаниям погруженной в реакционную среду термопары. Максимальное давление, которое достигалось в системе в процессе гидротермально-микроволновой обработки, составляло ~10 бар. После нагрева до 180°С и изотермической выдержки при этой температуре в течение указанного выше времени микроволновой нагрев отключали и проводили охлаждение стеклянной ампулы до 60°С (зависимости температуры от времени при гидротермально-микроволновой обработке приведены на рис. 1). Полученные осадки отделяли от маточного раствора отстаиванием (маточный раствор затем сливался), промывали дистиллированной водой и сушили при 80°C в течение 20 ч. Полученные таким образом девять образцов, различающихся продолжительностью изотермической выдержки при температуре 180°С, были подвергнуты дальнейшему анализу.
МЕТОДЫ ХАРАКТЕРИЗАЦИИ
Кристаллическую структуру полученных образцов исследовали методом рентгеновской дифракции с помощью порошкового рентгеновского дифрактометра Rigaku SmartLab 3 (Rigaku Corporation, Япония). Съемку рентгеновских дифрактограмм (излучение CoKα, Kβ-фильтр) выполняли в геометрии Брэгга–Брентано при комнатной температуре. Наблюдаемые параметры рефлексов (угол Брэгга (2θobs), полная ширина на полувысоте (FWHMobs) и интегральная ширина (βobs)) были определены в программном пакете Rigaku SmartLab Studio II. Коррекцию инструментального уширения (FWHMinstr) проводили с использованием снятого в аналогичных условиях эталона, представляющего собой монокристаллическую пластину SrTiO3 с полированной стороной, перпендикулярной кристаллографическому направлению [100]. Согласно критерию, определяющему тип профиля рефлекса [32], рефлексы во всех образцах являются псевдо-войтовскими, поэтому расчет скорректированного значения полной ширины на полувысоте (FWHMcorr) проводили с учетом псевдо-войтовской формы рефлексов (уравнение (1), [32]). Средние размеры кристаллитов (D) и микронапряжения (εs) были рассчитаны по методу Вильямсона–Холла для случая псевдо-войтовских рефлексов, наблюдаемых на дифрактограммах (уравнение (8), [33]). Расчет проводили по рефлексам с индексами Миллера 311, 222, 400, 440 и 622, принимая λ(CoKα1) = = 1.789001 Å, KScherrer = 0.94 и Kstrain = 4. Использование метода Вильямсона–Холла для расчета среднего размера кристаллитов оправдано присутствием во всех образцах микронапряжений и неправомерностью применения в этом случае традиционной формулы Шеррера (рис. S1 ).
Определение степени превращения (ξ) аморфной фазы в кристаллическую фазу BFWO проводили по данным рентгеновской дифракции путем сравнения интегральных интенсивностей аморфного гало и рефлексов от кристаллической фазы. При расчете массовой доли кристаллической фазы (степени превращения) параметр Хуанга был принят равным 1 (уравнение (3), [34]). Стоит, однако, отметить, что данный метод определения доли аморфной фазы в образце не является прецизионным, поскольку, во-первых, фон под аморфным гало проводится вручную, а во-вторых, коэффициент Хуанга, как правило, отличается от 1. Подобные приближения не позволяют уверенно полагаться на рассчитываемые таким образом значения доли аморфной фазы, но, тем не менее, это не мешает установить общую тенденцию ее изменения при анализе серии образцов.
Микрофотографии и валовый элементный состав образцов были получены на сканирующем электронном микроскопе Tescan Vega 3 SBH (Tescan Orsay Holding, Чешская Республика) с приставкой Oxford Instruments для рентгеноспектрального микроанализа (РСМА). Относительное количество элементов рассчитывали в программном пакете AZtec по следующим сериям электронных переходов: Fe по K-серии, Bi и W по L- и M-сериям. Спектры характеристического излучения каждого образца накапливались с трех участков общей площадью ~9 мм2 с набором статистики не менее 2.5 млн импульсов на каждом участке. Полученные с каждого участка показания усредняли, а погрешность измерений определяли для доверительной вероятности 0.95.
Исследования методом просвечивающей электронной микроскопии (ПЭМ) проводили с использованием микроскопа JEM-2100F при ускоряющем напряжении 200 кВ. Пробы образцов были подготовлены одним из традиционных способов: диспергирование взвеси навески порошка в этиловом спирте ультразвуком в течение 8–10 мин с последующим осаждением и сушкой на тонкой поддерживающей углеродной пленке.
Спектры оптического поглощения (или диффузного отражения в интегрирующей сфере в случае непрозрачных образцов) были получены при комнатной температуре с помощью спектрофотометра Cary Varian 5000 в диапазоне от 0.6 до 5.0 эВ. Определение оптической ширины запрещенной зоны проводили по методу Тауца [35]:
(1)
${{\left( {h\nu F\left( r \right)} \right)}^{{{1 \mathord{\left/ {\vphantom {1 n}} \right. \kern-0em} n}}}} = A\left( {h\nu - {{E}_{{\text{g}}}}} \right),$РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Химический состав полученных образцов
Данные по валовому химическому составу образцов, полученные методом РСМА, представлены в табл. 1. Валовый состав всех образцов в целом хорошо соответствует номинальному составу, заложенному по синтезу. Полученные результаты подтверждают установленные ранее закономерности, в соответствии с которыми в системе Bi2O3–Fe2O3–WO3 состав образующейся в гидротермальных условиях фазы пирохлора совпадает с номинальным составом, если pH среды гидротермального флюида находится в диапазоне от ~2 до ~5 [31]. Некоторые отличия валового состава образцов могут быть обусловлены как синтетическими ошибками, так и ограниченной точностью используемого метода анализа состава. Кроме основных элементов (Bi, Fe, W и O) почти во всех полученных образцах на пределе чувствительности метода РСМА фиксируется незначительное количество примеси натрия, что, по-видимому, обусловлено неполной отмывкой порошка от маточного раствора.
Таблица 1.
Валовый элементный состав образцов, синтезированных при различной продолжительности изотермической выдержки, по данным РСМА
| Продолжительность изотермической выдержки (τ) | Валовый элементный состав, отн. ат. ед. | ||
|---|---|---|---|
| Bi/W ± Δ | Fe/W ± Δ | Bi/Fe ± Δ | |
| 1 с | 0.56 ± 0.01 | 0.37 ± 0.02 | 1.53 ± 0.05 |
| 15 с | 0.50 ± 0.08 | 0.37 ± 0.02 | 1.35 ± 0.15 |
| 30 с | 0.56 ± 0.05 | 0.37 ± 0.02 | 1.49 ± 0.07 |
| 45 с | 0.55 ± 0.03 | 0.37 ± 0.02 | 1.47 ± 0.03 |
| 1 мин | 0.53 ± 0.02 | 0.37 ± 0.01 | 1.44 ± 0.04 |
| 2 мин | 0.52 ± 0.04 | 0.37 ± 0.02 | 1.39 ± 0.06 |
| 3 мин | 0.52 ± 0.03 | 0.38 ± 0.02 | 1.39 ± 0.08 |
| 4 мин | 0.52 ± 0.09 | 0.37 ± 0.03 | 1.39 ± 0.12 |
| 5 мин | 0.48 ± 0.03 | 0.37 ± 0.01 | 1.29 ± 0.07 |
Порошковая рентгеновская дифракция полученных образцов
Порошковые рентгеновские дифрактограммы полученных образцов представлены на рис. 2а. Из кристаллических фаз на дифрактограммах присутствует только фаза со структурой кубического пирохлора (CSD 1961005, [30]), поскольку положения всех имеющихся брэгговских рефлексов полностью совпадают с рефлексами этой фазы. Параметр a элементарной ячейки фазы BFWO практически не меняется при изменении продолжительности изотермической выдержки и составляет ~10.34 Å для всех полученных образцов.
Рис. 2.
Рентгеновские дифрактограммы образцов, синтезированных при различной продолжительности (τ) изотермической (180°С) выдержки (а); зависимости степени превращения (ξ) аморфной фазы в кристаллическую BFWO (правая ось ординат) и среднего размера (D) кристаллитов фазы BFWO (левая ось ординат) от продолжительности изотермической выдержки (б).
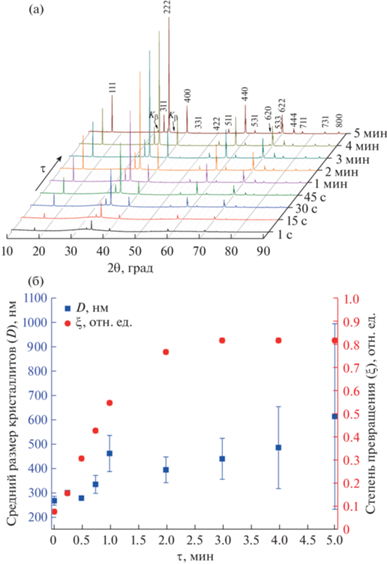
Визуальный качественный анализ указывает на присутствие на дифрактограммах помимо узких брэгговских рефлексов широкого гало, регистрирующегося в результате безинтерференционного рассеяния излучения аморфной фазой, не имеющей дальнего порядка. По мере увеличения продолжительности изотермической выдержки растет интегральная интенсивность брэгговских рефлексов фазы BFWO и уменьшается интегральная интенсивность аморфного гало. Для количественной оценки степени превращения аморфной фазы в кристаллическую BFWO было проведено сравнение интегральных интенсивностей аморфного гало и рефлексов от кристаллической фазы BFWO. Расчет средних размеров кристаллитов был проведен методом Вильямсона–Холла (из-за присутствующих в кристаллитах микродеформаций, что хорошо видно на графиках (рис. S1 )) для рефлексов с псевдо-войтовскими профилями.
В результате расчета была получена зависимость степени превращения ξ от продолжительности изотермической выдержки (рис. 2б, правая ось ординат). С этой зависимостью коррелирует зависимость среднего размера кристаллитов D фазы BFWO от продолжительности изотермической выдержки (рис. 2б, левая ось ординат), которая четко показывает тенденцию роста размера кристаллитов с увеличением продолжительности изотермической выдержки (несмотря на большие стандартные отклонения δD), особенно для образцов с большой продолжительностью изотермической выдержки и, соответственно, большим размером кристаллитов (большие значения δD возникают из-за большого разброса экспериментальных точек на графике Вильямсона–Холла, очевидно, из-за трудности прецизионного определения FWHMobs узких рефлексов).
При увеличении продолжительности изотермической выдержки от 1 с до 2 мин в образцах наблюдается заметный рост степени превращения и среднего размера кристаллитов, который при дальнейшем увеличении ее продолжительности с 2 до 5 мин замедляется. Поскольку в образце, полученном при изотермической выдержке 1 с, средний размер кристаллитов уже составляет ~270 нм, можно предположить, что процесс формирования фазы BFWO протекает при гораздо более низкой температуре, чем температура изотермического режима синтеза, равная 180°C (рис. 1).
Представленные на рис. 2б зависимости указывают на то, что строительным материалом для формирования кристаллитов фазы BFWO является аморфная фаза близкого к ней состава. Последнее положение подтверждается тем, что валовый состав всех образцов практически одинаковый (табл. 1), т.е. не зависит от массового соотношения кристаллической и аморфной фаз в образце. В пользу этого предположения говорит и наличие микродеформаций в кристаллитах (рис. S1 ), которые, возможно, возникают из-за наличия в них дефектов и соприкосновения их с окружающей аморфной фазой близкого состава и структуры (на локальном уровне).
Сканирующая электронная микроскопия полученных образцов
Микрофотографии образцов, полученных при изотермической выдержке в течение 1, 30 с и 1, 5 мин, представлены на рис. 3 (15, 45 с и 2, 3, 4 мин на рис. S2 ). На микрофотографиях образцов, полученных при 1 и 30 с изотермической выдержки (рис. 3а и 3б соответственно), можно выделить редкие частицы размером ~350‒450 нм, форма которых напоминает плохо ограненный куб или сферу. С увеличением продолжительности изотермической выдержки количество таких частиц в образцах растет, и можно отметить, что в случае образца, полученного при изотермической выдержке в течение 5 мин (рис. 3г), почти весь массив частиц представлен крупными октаэдрическими частицами. Помимо крупных частиц в образцах, полученных при малой продолжительности изотермической выдержки, присутствует большое количество частиц, размер которых равен ~100 нм (рис. 3а, 3б).
Рис. 3.
Изображения СЭМ, полученные от образцов, синтезированных при различной продолжительности изотермической выдержки: 1 (а), 30 с (б) и 1 (в), 5 мин (г).

Количество таких маленьких частиц постепенно уменьшается с увеличением продолжительности изотермической выдержки, и в образце, полученном при 5 мин изотермической выдержки, такие частицы присутствуют уже в незначительном количестве (рис. 3г). Средний размер кристаллитов, рассчитанный по данным рентгеновской дифракции, в образце, полученном при 1 с изотермической выдержки, уже составляет ~270 нм и постепенно расчет с увеличением ее продолжительности (рис. 2б, левая ось ординат). Таким образом, можно сделать вывод, что кристаллическая фаза в образцах представлена именно крупными частицами, габитус которых в конечном счете приобретает форму октаэдра. Малые же частицы, по всей видимости, имеют аморфную структуру и являются агломератами. Данный вывод соответствует характеру изменения степени превращения и среднего размера кристаллитов фазы BFWO с ростом продолжительности изотермической выдержки (рис. 2б), поскольку доля и размер крупных кристаллических частиц постепенно увеличиваются с ростом продолжительности изотермической выдержки, а доля малых аморфных частиц уменьшается.
Просвечивающая электронная микроскопия полученных образцов
Данные ПЭМ для образцов, полученных при продолжительности изотермической выдержки 1, 30 с и 5 мин, представлены на рис. 4а, 4б и 4в соответственно. В образце, полученном при 1 с изотермической выдержки (рис. 4а), можно выделить два морфологических мотива: крупные частицы (~300‒450 нм), плохо пропускающие первичный электронный пучок, и массив из более мелких частиц, собранных в плоские агломераты, распределенные по подложке. На электронной дифрактограмме, полученной с некоторой области таких маленьких частиц, можно выделить широкое кольцо малого радиуса, находящееся вблизи первичного пучка, а также два узких кольца, относящихся к дифракции от подложки (углеродной пленки, поддерживающей частицы, рис. S3 ). Уширенное кольцо на электронной дифрактограмме свидетельствует о наличии в образце вещества, в котором отсутствует дальний порядок.
Рис. 4.
Изображения ПЭМ, полученные от образцов, синтезированных при различной продолжительности изотермической выдержки: 1 (а), 30 с (б) и 5 мин (в).
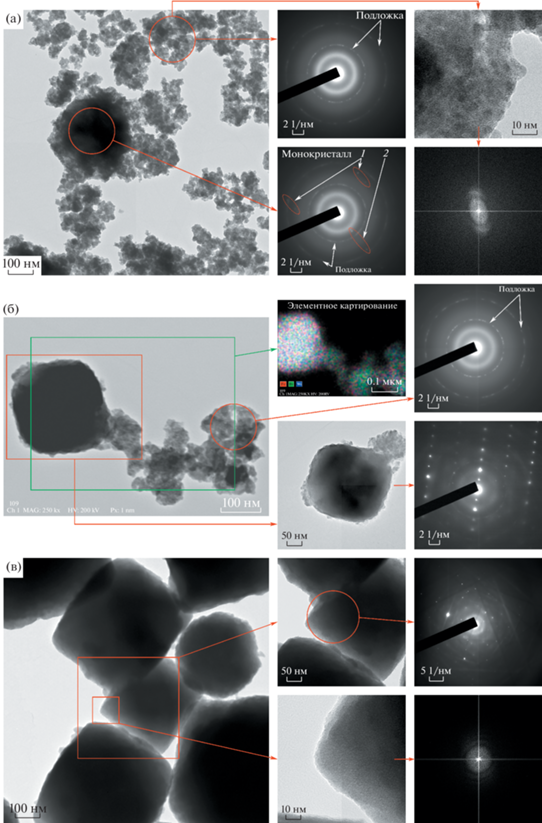
При исследовании таких частиц при высоких увеличениях в режиме разрешения решетки (HRTEM) не удалось увидеть никаких признаков кристаллических плоскостей (на live-FFT ни разу не появлялись точечные рефлексы). Это также говорит об аморфности таких малых частиц.
В то же время на дифракционной картине, полученной с ограниченной области крупной частицы (рис. 4а), можно увидеть признаки монокристалла, а именно точечные рефлексы, отстоящие друг от друга на вектор трансляции обратной решетки. Помимо точечных рефлексов на дифракционной картине наблюдается уширенное кольцо от аморфной фазы. По всей видимости, это свидетельствует о том, что крупные частицы имеют структуру ядро–оболочка, где в качестве ядра выступают кристаллы фазы BFWO, а оболочку образует слой аморфной фазы.
Данные ПЭМ для образца, полученного при изотермической выдержке в течение 30 с, представлены на рис. 4б. В этом образце также можно выделить два морфологических мотива: крупные частицы (~300‒450 нм), габитус которых напоминает плохо ограненный октаэдр, и плоские агломераты, состоящие из частиц значительно меньших размеров. Наблюдаемая дифракционная картина, полученная со скопления таких маленьких частиц, характерна для аморфной фазы. На дифракционной картине, полученной с крупной частицы, присутствуют ряды точечных рефлексов от монокристалла, образующего ядро частицы, а также тусклое уширенное кольцо малого радиуса от аморфной фазы, формирующей оболочку частицы. В данном случае ядро частицы ориентировано к первичному пучку таким образом, что одна из систем плоскостей находится в брэгговском положении, что видно по появлению на дифракционной картине рядов точечных рефлексов. Основными векторами трансляции на представленной дифракционной картине, по всей видимости, являются g = (111) и (331), которым соответствуют измеренные межплоскостные расстояния 6.1 и 2.4 Å соответственно.
Результаты элементного (Bi, Fe и W) картирования поверхности крупных и малых частиц свидетельствуют о том, что распределение всех элементов в выделенной области является равномерным, причем визуально не обнаруживается отличий в распределении элементов между двумя видами частиц (рис. 4б).
В образце, полученном при изотермической выдержке в течение 5 мин (рис. 4в), преобладающим морфологическим мотивом являются крупные частицы (~0.35‒0.8 мкм), большинство которых имеет форму октаэдра, в то время как агломераты малых частиц аморфной фазы встречаются на поверхности подложки значительно реже. Представленная дифракционная картина с ограниченной области крупной частицы является классическим примером дифракции от большого монокристалла, поскольку помимо рядов точечных рефлексов отчетливо видны линии Кикучи ‒ темные и светлые полосы. Линии Кикучи, возникающие на дифракционной картине, являются следствием диффузионного рассеяния электронов в толще монокристалла. Таким образом, их появление на ПЭМ-дифрактограммах возможно только при дифракции от довольно толстого монокристалла, для которого кинематическое приближение не применимо. Помимо точечных рефлексов и линий Кикучи на дифракционной картине обнаруживается тусклое уширенное кольцо малого радиуса, свидетельствующее о присутствии аморфной фазы. При исследовании тонких краев таких частиц при высоких увеличениях в режиме разрешения решетки (HRTEM), как и в случае образцов с меньшей продолжительностью изотермической выдержки, не удалось увидеть никаких признаков кристаллических плоскостей. Это также свидетельствует об аморфной структуре оболочки.
Представленные результаты говорят о том, что при продолжительности изотермической выдержки порядка нескольких минут почти вся масса аморфной фазы располагается в оболочке композиционных частиц, ядро которых составляют кристаллы фазы BFWO. Массовая доля аморфной фазы в таких образцах составляет ~20% (рис. 2б), откуда можно заключить, что аморфная фаза образует вокруг ядра довольно толстую оболочку. Массовое соотношение ядра и оболочки отдельно взятой частицы будет определять ее габитус. Если в массовом соотношении преобладает оболочка, то форма частиц напоминает куб или сферу, что хорошо заметно в случае образцов, полученных при продолжительности изотермической выдержки <1 мин. Если же масса ядра больше массы аморфной оболочки, то форма частиц похожа на октаэдр (по всей видимости, это габитус монокристалла или их сростка ‒ ядра), что наблюдается для образцов, полученных при продолжительности изотермической выдержки >1 мин.
Оптические свойства полученных образцов
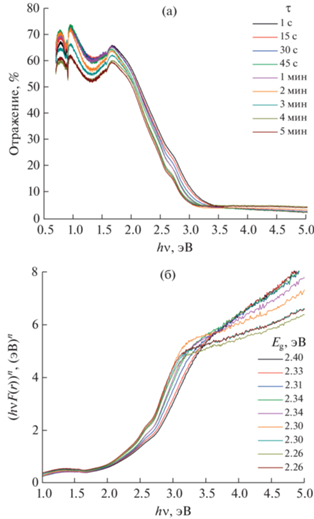
Спектры диффузного отражения образцов представлены на рис. 5а, а рассчитанные по ним кривые Тауца построены на рис. 5б. Значения Eg полученных образцов для прямых разрешенных электронных переходов, определенные по пересечению оси hν касательными к прямолинейным участкам кривых Тауца, приведены в описании к рис. 5б. Представленные значения Eg полученных композиционных материалов находятся в диапазоне 2.26‒2.40 эВ, что характеризует эти материалы как широкозонные полупроводники. В целом можно отметить тенденцию к уменьшению Eg образцов при увеличении продолжительности изотермической выдержки гидротермального флюида в процессе их синтеза, что естественным образом приводит к постепенному переходу их светлого оранжевого оттенка к более темному. Полученные значения Eg являются интегральными, поскольку все образцы двухфазные. Тем не менее можно отметить, что Eg кристаллической фазы BFWO должна быть заметно меньше, чем аморфной фазы, поскольку при увеличении массовой доли кристаллической фазы интегральное значение Eg композита уменьшается. Поскольку химический состав образцов практически не меняется, а размеры кристаллитов слишком велики для активации размерных эффектов, по всей видимости, именно изменение массового соотношения сосуществующих фаз приводит к изменению интегрального значения Eg полученных композиционных материалов. Этот результат можно использовать для целенаправленного синтеза функциональных материалов с заданными оптическими характеристиками с целью их применения в фотокатализе и фотовольтаике.
ЗАКЛЮЧЕНИЕ
Исследован процесс формирования фазы BFWO в условиях гидротермально-микроволновой обработки при продолжительности изотермической (180°C) выдержки гидротермального флюида от 1 с до 5 мин. Показано образование композитов состава BFWO/аморфная фаза, определены их химический состав, морфология, размер частиц и кристаллитов. Установлена корреляция между зависимостью степени превращения аморфной фазы в кристаллическую и зависимостью среднего размера кристаллитов фазы BFWO от продолжительности изотермической выдержки, а именно: обе величины возрастают с увеличением продолжительности изотермической выдержки. Выявлено, что формирование фазы BFWO начинается еще до достижения температуры изотермической выдержки, поскольку даже при ее продолжительности в 1 с средний размер кристаллитов уже составляет ~270 нм.
Показано формирование частиц типа ядро–оболочка, в которых ядро частицы образуют кристаллы фазы BFWO, а оболочку – слой аморфной фазы. Установлено, что с ростом степени превращения и среднего размера кристаллов фазы BFWO габитус таких частиц все больше приобретает форму октаэдра, которую, по-видимому, имеют ядра частиц, образованные монокристаллами или их сростками. Показано, что аморфная фаза, имея схожий химический состав, является строительным материалом для формирования кристаллической фазы BFWO.
Определены значения Eg (2.26‒2.40 эВ) для прямых разрешенных электронных переходов в зависимости от соотношения сосуществующих в композиционных материалах фаз (BFWO/аморфная фаза). Показано уменьшение Eg при увеличении в композите доли фазы BFWO, что дает возможность варьировать Eg получаемых композитов, меняя степень превращения аморфной фазы в кристаллическую.
Список литературы
Jitta R.R., Gundeboina R., Veldurthi N.K. et al. // J. Chem. Technol. Biotechnol. 2015. V. 90. № 11. P. 1937. https://doi.org/10.1002/jctb.4745
Anantharaman A.P., Dasari H.P. // Ceram. Int. 2021. V. 47. № 4. P. 4367. https://doi.org/10.1016/j.ceramint.2020.10.012
Gardner J.S., Gingras M.J.P., Greedan J.E. // Rev. Mod. Phys. 2010. V. 82. № 1. P. 53. https://doi.org/10.1103/revmodphys.82.53
Wiebe C.R., Hallas A.M. // APL Mater. 2015. V. 3. № 4. Art. 041519. https://doi.org/10.1063/1.4916020
Piir I.V., Koroleva M.S., Ryabkov Y.I. et al. // J. Solid State Chem. 2013. V. 204. P. 245. https://doi.org/10.1016/j.jssc.2013.05.031
Sun S., Liu Z., Ti R. et al. // Ceram. Int. 2021. V. 47. № 5. P. 6049. https://doi.org/10.1016/j.ceramint.2020.10.180
Dasin N.A.M., Tan K.B., Khaw C.C. et al. // Ceram. Int. 2017. V. 43. № 13. P. 10183. https://doi.org/10.1016/j.ceramint.2017.05.043
Babu G.S., Bedanta S., Valant M. // Solid State Commun. 2013. V. 158. P. 51. https://doi.org/10.1016/j.ssc.2012.11.025
Ellert O.G., Egorysheva A.V., Gajtko O.M. et al. // J. Magn. Magn. Mater. 2018. V. 463. P. 13. https://doi.org/10.1016/j.jmmm.2018.05.035
Sun S., Zhu L., Zhang B. et al. // J. Mater. Chem. C. 2019. V. 7. № 5. P. 1263. https://doi.org/10.1039/c8tc05672b
Radha R., Ravi Kumar Y., Sakar M. et al. // Appl. Catal. B. 2018. V. 225. P. 386. https://doi.org/10.1016/j.apcatb.2017.12.004
Egorysheva A.V., Gajtko O.M., Rudnev P.O. et al. // Eur. J. Inorg. Chem. 2016. № 13–14. P. 2193. https://doi.org/10.1002/ejic.201501159
Bencina M., Valant M., Pitcher M.W. et al. // Nanoscale. 2014. V. 6. № 2. P. 745. https://doi.org/10.1039/c3nr04260j
Samu G.F., Veres Á., Endrődi B. et al. // Appl. Catal. B. 2017. V. 208. P. 148. https://doi.org/10.1016/j.apcatb.2017.02.036
Lomakin M.S., Proskurina O.V., Sergeev A.A. et al. // J. Alloys Compd. 2021. V. 889. 161598. https://doi.org/10.1016/j.jallcom.2021.161598
Michael E.K., Trolier-McKinstry S. // J. Appl. Phys. 2015. V. 118. № 5. Art. 054101. https://doi.org/10.1063/1.4927738
Martínez-Coronado R., Alonso J.A., Cascos V. et al. // J. Power Sources. 2014. V. 247. P. 876. https://doi.org/10.1016/j.jpowsour.2013.08.125
Scharnberg A.R. de A., Berutti F.A., Alves A.K. // Environ. Technol. 2020. https://doi.org/10.1080/09593330.2020.1758218
Allured B., DelaCruz S., Darling T. et al. // Appl. Catal. B. 2014. V. 144. P. 261. https://doi.org/10.1016/j.apcatb.2013.07.019
Vanderah T.A., Levin I., Lufaso M.W. // Eur. J. Inorg. Chem. 2005. V. 14. P. 2895. https://doi.org/10.1002/ejic.200500234
Jusoh F.A., Tan K.B., Zainal Z. et al. // J. Mater. Res. Technol. 2020. V. 9. № 5. P. 11022. https://doi.org/10.1016/j.jmrt.2020.07.102
Zhuk N.A., Krzhizhanovskaya M.G., Koroleva A.V. et al. // Inorg. Chem. 2021. V. 60 № 7. P. 4924. https://doi.org/10.1021/acs.inorgchem.1c00007
Krasnov A.G., Piir I.V., Sekushin N.A. et al. // Russ. J. Electrochem. 2017. V. 53. № 8. P. 866. https://doi.org/10.1134/S1023193517080122
Chon M.P., Tan K.B., Khaw C.C. et al. // J. Alloys Compd. 2016. V. 675. P. 116. https://doi.org/10.1016/j.jallcom.2016.03.089
Khaw C.C., Tan K.B., Lee C.K. et al. // J. Eur. Ceram. Soc. 2012. V. 32. № 3. P. 671. https://doi.org/10.1016/j.jeurceramsoc.2011.10.012
Valant M., Babu G.S., Vrcon M. et al. // J. Am. Ceram. Soc. 2011. V. 95. № 2. P. 644. https://doi.org/10.1111/j.1551-2916.2011.04801.x
Kuvshinova T.B., Egorysheva A.V., Gaitko O.M. et al. // Russ. J. Inorg. Chem. 2015. V. 60. № 10. P. 1179. https://doi.org/10.1134/S0036023615100113
Modeshia D.R., Walton R.I. // Chem. Soc. Rev. 2010. V. 39. № 11. P. 4303. https://doi.org/10.1039/b904702f
Daniels L.M., Playford H.Y., Grenèche J.-M. et al. // Inorg. Chem. 2014. V. 53. № 24. P. 13197. https://doi.org/10.1021/ic502411z
Lomakin M.S., Proskurina O.V., Danilovich D.P. et al. // J. Solid State Chem. 2020. V. 282. Art. 121064. https://doi.org/10.1016/j.jssc.2019.121064
Lomakin M.S., Proskurina O.V., Gusarov V.V. // Nanosyst.: Phys. Chem. Math. 2020. V. 11. № 2. P. 246. https://doi.org/10.17586/2220-8054-2020-11-2-246-251
Terlan B., Levin A.A., Börrnert F. et al. // Eur. J. Inorg. Chem. 2016. V. 21. P. 3460. https://doi.org/10.1002/ejic.201600315
Terlan B., Levin A.A., Börrnert F. et al. // Chem. Mater. 2015. V. 27. № 14. P. 5106. https://doi.org/10.1021/acs.chemmater.5b01856
Abrosimova G.E., Aronin A.S., Kholstinina N.N. // Phys. Solid State. 2010. V. 52. № 3. P. 445. https://doi.org/10.1134/s1063783410030017
Бонч-Бруевич В.Л., Калашников С.Г. Физика полупроводников. М.: Мир, 1977. 678 с.
Кортюм Г., Браун В., Герцог Г. // Успехи физ. наук. 1965. Т. 85. № 2. С. 365.
Дополнительные материалы
- скачать ESM.docx
- Рис. S1. Графики Вильямсона-Холла для образцов, синтезированных при различной продолжительности изотермической выдержки (1 (а), 15 (б), 30 (в), 45 (г) сек и 1 (д), 2 (е), 3 (ё), 4 (ж), 5 (з) мин)
Рис. S2. Изображения СЭМ, полученные от образцов, синтезированных при различной продолжительности изотермической выдержки (15 (а), 45 (б) сек и 2 (в), 3 (г), 4 (д) мин)
Рис. S3. Дифракционная картина, наблюдаемая от подложки
Инструменты
Журнал неорганической химии