

Журнал неорганической химии, 2022, T. 67, № 2, стр. 241-247
О гетеробиядерных комплексах Cu2+ и Zn2+ на основе глутатионатных комплексов золота(I) в водном растворе
И. В. Миронов a, *, В. Ю. Харламова a
a Институт неорганической химии им. А.В. Николаева СО РАН
630090 Новосибирск, пр-т Академика Лаврентьева, 3, Россия
* E-mail: imir@niic.nsc.ru
Поступила в редакцию 24.06.2021
После доработки 11.09.2021
Принята к публикации 13.09.2021
- EDN: PBYYPC
- DOI: 10.31857/S0044457X2202009X
Аннотация
Исследовано взаимодействие Cu2+ и Zn2+ с высокоустойчивыми глутатионатными комплексами золота(I) в водном растворе (t = 25°C, I = 0.2 M (NaCl)), приводящее к образованию гетеробиядерных комплексов, в которых глутатионат координирован к золоту(I) через депротонированную тиольную группу, а медь(II) или цинк(II) связаны с глицинатными фрагментами (ГФ) глутатиона. Показано, что в растворе медь(II) связывается с двумя ГФ, относящимися к разным глутатионат-ионам, а цинк(II) – с одним ГФ. В области pH 6–9 для полимерного (1 : 1) комплекса (AuGS)m${\text{H}}_{m}^{{m--}}$ наиболее вероятными формами гетеробиядерных комплексов являются (AuGS)mCun${\text{H}}_{{m - 2n}}^{{m--}}$ и (AuGS)m(ZnOH)n${\text{H}}_{{m - n}}^{{m--}}$ с переменным количеством M2+, где GS3– – депротонированный остаток глутатиона. Растворы остаются гомогенными пока $C_{{\text{M}}}^{{2 + }}$ : CGS < 0.5. При $C_{{\text{M}}}^{{2 + }}$ : CGS > 0.5 наблюдается образование твердых фаз комплексов.
ВВЕДЕНИЕ
Многоядерные комплексы, в структуре которых одновременно присутствуют два или несколько центральных атомов-комплексообразователей, играют важную роль в современной химии. Такие соединения перспективны в качестве противоопухолевых препаратов, контрастирующих агентов для МРТ, основы для получения биметаллических наночастиц [1–3]. Так, гетеробиядерные комплексы золота(I) и платины(II), а также золота(I) и рутения(II) с некоторыми лигандами проявляют антипролиферативные свойства [4, 5]. Присутствие различных металлических центров в одной молекуле может приводить к усилению цитотоксических эффектов комплексов.
Комплексы золота(I) с анионами тиолсодержащих кислот имеют практическое применение. Например, их часто используют для функционализации наночастиц [6, 7], и в ряде работ [8–10] показано, что получаемые системы обладают сильной флуоресценцией. Кроме того, эти комплексы применяют в медицине [11–13]. Так, комплекс золота(I) с тиомалатом (миокризин) используют в терапии ревматоидного артрита. Комплексы золота(I) с глутатионом и цистеином образуются в организме при использовании соединений золота(III) и золота(I) в качестве антираковых средств. Анионы тиолсодержащих кислот координированы к золоту(I) через атом S тиольной группы, и такие комплексы обладают очень высокой устойчивостью. В то же время в состав этих лигандов часто входят и другие группы: амино- (–NH2) и карбоксильные (–COO–), не занятые в координации к золоту(I), но способные присоединять ионы других металлов. Из-за возможности образования хелатов особенно перспективна в этом отношении группа NH2–CH(R)–COO–, входящая в состав остатков α-аминокислот и некоторых пептидов, в частности, цистеина и глутатиона. В настоящей работе рассмотрены гетеробиядерные комплексы на основе глутатионатов золота(I), содержащие дополнительно медь(II) или цинк(II).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В работе использовали раствор HAuCl4 [14], хлористый натрий (ос. ч.), соляную кислоту (фиксанал), безводный сульфит натрия (ч. д. а.), L-глутатион восстановленный (АО “Вектон”, Россия, >98%), раствор NaOH (“без CO2”), прокипяченную бидистиллированную воду. Концентрацию HAuCl4 устанавливали по УФ-поглощению раствора (ε = 5600 M–1 см–1 при 314 нм, среда 0.1 M HCl).
Все эксперименты проводили при 25°C (водяной термостат U7) и I = 0.2 M (NaCl). Раствор Na2SO3 (C = 0.2 моль/л) готовили непосредственно перед экспериментом из безводного реактива.
Рабочие растворы готовили, восстанавливая ${\text{AuCl}}_{4}^{ - }$ до ${\text{AuCl}}_{2}^{ - }$ (CAu = (1–10) × 10–3 моль/л) сульфитом натрия (${\text{AuCl}}_{4}^{ - }$ + ${\text{SO}}_{3}^{{2 - }}$ + H2O = ${\text{AuCl}}_{2}^{ - }$ + + ${\text{SO}}_{4}^{{2 - }}$ + 2H+ + 2Cl–) в присутствии NaCl (0.2 моль/л) и добавки NaOH (nNaOH/nAu = 3.00). Для ускорения процесса возможен небольшой подогрев раствора (<30°C). Получаемый раствор обычно имеет pH 5–6 и является метастабильным к диспропорционированию: ${\text{3AuCl}}_{2}^{ - }$ = 2Au0 + + ${\text{AuCl}}_{4}^{ - }$ + 2Cl– [15]. По окончании восстановления (20 мин) к раствору добавляли требуемую навеску глутатиона (GSH3). Общий объем составлял 30–50 мл.
Помимо расхода на восстановление ${\text{AuCl}}_{4}^{ - },$ сульфит-ион ${\text{SO}}_{3}^{{2 - }}$ окисляется растворенным кислородом, особенно быстро в щелочной области. Использование прокипяченной воды снижает влияние кислорода, но не устраняет его полностью. Поэтому в работе не использовали низкие (<10–3 моль/л) концентрации компонентов, а Na2SO3 для восстановления брали в небольшом (до 10%, подбирается эмпирически) избытке [16]. Использование других мер нежелательно. Так, пропускание в раствор инертного газа часто приводит к снятию метастабильности ${\text{AuCl}}_{2}^{ - }$ и выделению золота(0). Систематических ошибок, связанных с окислением кислородом, в ходе экспериментов не выявлено.
Измерения pH проводили при помощи стеклянного комбинированного электрода ЭСК 10301/7, прибор Radelkis OP-208. Время установления потенциала составляло 2–3 мин. Электрод калибровали по растворам сильной кислоты HCl в 0.2 M NaCl, т.е. измеряемые величины рН равны –lg[H+]. Необходимое для расчетов ионное произведение воды для 25°С и 0.2 M NaCl равно lgKw = 13.76 [17].
Спектры поглощения записывали на спектрофотометре Genesys 6 (Thermo Spectronic) в диапазоне длин волн 400–800 нм, l = 0.05–5 см, раствор сравнения – вода. Проверку способности образцов растворов к флуоресценции проводили на спектрофлуориметре Agilent Cary Eclipse.
ИК-спектры регистрировали на фурье-спектрометре Scimitar FTS 2000 (Digilab) при ν = 400–4000 см–1. Образцы спрессовывали с сухим KBr под вакуумом.
Осадки для анализа отделяли фильтрованием через стеклянный фильтр. Для удаления хлорида их 4 раза промывали водой порциями по 5 мл и после двукратной промывки спиртом сушили на воздухе. Элементный CHNS-анализ проводили в аналитической лаборатории ИНХ СО РАН на CHNS-анализаторе vario MICRO cube (Elementar). Для Au(GSH)Cu · 0.5SO4 · 1.5H2O (% опр./расч.): N (6.6/6.6); C (18.9/18.7); H (2.8/2.8); S (7.1/7.5).
Количественный анализ на медь проводили спектрофотометрически аммиачным методом после окисления пробы азотной кислотой при нагревании. Концентрацию меди определяли по поглощению аммиачных комплексов в среде 2 M NH3. Анализ на золото проводили спектрофотометрически в форме ${\text{AuCl}}_{4}^{ - }$ после разложения пробы царской водкой и удаления азотной кислоты.
Оценку эффективной константы $K_{{\text{H}}}^{*}$ выполняли при помощи нелинейного МНК [18]. Расчет других величин описан в тексте ниже. В качестве ошибок указаны стандартные отклонения.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
может присоединять четыре иона H+ и имеет следующие константы протонирования lgKHi: 9.58 (i = 1), 8.76 (i = 2), 3.58 (i = 3), 2.58 (i = 4) [18]. Первые две относятся к протонированию тиольной (–S–) и амино- (–NH2) групп, третья и четвертая – к протонированию карбоксильных (–COO–) групп. Тиольная и аминогруппы протонированы уже в щелочной области, карбоксильные – только в кислой. Концентрации форм зависят от pH раствора. Далее набор форм разной степени протонирования мы будем обозначать GS*, т.е. [GS*] = = $\Sigma \left[ {{\text{GSH}}_{i}^{{i--3}}} \right].$ К золоту(I) глутатионат координируется через депротонированную S–-группу, остальные группы в координации не участвуют, хотя остаются способными к протонированию и координации к другим ионам металлов.
Золото(I) способно образовывать с GS* два вида комплексов: полимерные $({\text{AuGS}})_{m}^{*},$ в которых атомы серы являются мостиковыми, и мономерные ${\text{Au(GS}})_{2}^{*}$ [18]. Однако степень полимеризации (m) и вид (циклические или линейные) не определены. Как и GS3–, все комплексы способны к протонированию за счет амино- и карбоксильных групп, т.е. $\left[ {({\text{AuGS}})_{m}^{*}} \right]$ = = Σ[(AuGS)m${\text{H}}_{i}^{{i--2m}}$]. Других комплексов, кроме указанных здесь с соотношением Au : GS = 1 : 1 и 1 : 2, не выявлено [18]. Равновесие
(1)
$1{\text{/}}m\left( {{\text{AuGS}}} \right)_{m}^{*} + {\text{ GS}}* = {\text{Au}}\left( {{\text{GS}}} \right)_{2}^{*}$При прямом восстановлении ${\text{AuCl}}_{4}^{ - }$ глутатионом в растворе образуется неопределенный набор его окисленных форм. Поэтому в своих экспериментах мы вначале получали комплекс золота(I) ${\text{AuCl}}_{2}^{ - }$ восстановлением ${\text{AuCl}}_{4}^{ - }$ сульфитом натрия и затем уже добавляли глутатион и NaOH до нужных значений CGS : CAu и pH. Также раствор содержал 0.2 M NaCl во избежание быстрого диспропорционирования: 3AuCl2– ↔ 2Au0 + ${\text{AuCl}}_{4}^{ - }$ + + 2Cl–.
Как отмечено выше, в состав GS* и комплексов золота(I) с GS* входит глицинатный фрагмент (ГФ) – группа NH2–CH(R)–COO–, способная к координации с образованием хелата с ионами переходных металлов. В области pH 6–9 аминогруппа находится в протонированном состоянии, а карбоксильные – в депротонированном. На рис. 1 приведены кривые титрования щелочью растворов, содержащих $({\text{AuGS}})_{m}^{*}$ и ${\text{Au}}({\text{GS}})_{2}^{*},$ с различным соотношением CGS/CAu в координатах COH/CGS–pH. При CGS : CAu ≤ 1.5 они практически совпадают. Здесь же показана область существования осадка (AuGSH2)m. Таким образом, при pH > 5.5 комплексы золота(I) в растворе содержат глутатион с депротонированными COO–-группами и протонированной аминогруппой $--{\text{NH}}_{3}^{ + }.$ Дальнейшее добавление щелочи приводит к ее постепенному депротонированию. Вследствие высокой устойчивости комплексов в этих условиях концентрация свободного GS* очень мала. Оценка эффективной константы протонирования NH2-группы глутатиона в комплексах [18], полученная на основании данных pH-метрического титрования (pH > 5.5), приводит к величине $\lg K_{{\text{H}}}^{*}$ = 9.3, которая выше, чем константа lgK2H свободного глутатиона. Вероятно, это вызвано более высоким общим отрицательным зарядом комплексов по сравнению с GSH2–, а также отличием K2H от детальной (“микроскопической”) константы [19] протонирования NH2-группы в глутатионе вследствие влияния тиольной группы. Из совпадения кривых титрования (рис. 1) следует, что при известном значении pH величину $K_{{\text{H}}}^{*}$ можно использовать для расчета концентрации ионов H+, связанных с NH2-группами глутатиона в комплексах:
(2)
${{C}_{{{\text{H}}\,\,{\text{связ}}}}} = n*{\kern 1pt} {{C}_{{{\text{GS}}}}},\,\,\,\,n{\kern 1pt} * = K_{{\text{H}}}^{*}[{{{\text{H}}}^{ + }}]{\text{/}}(1 + K_{{\text{H}}}^{*}[{{{\text{H}}}^{ + }}]).~$Рис. 1.
Кривые титрования щелочью растворов с различным соотношением GS : Au: 1 – 1 : 1; 2 – 1.25 : 1; 3 – 1.5 : 1.
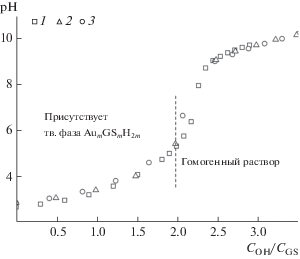
Очевидно, эту же величину CH связ можно определять и прямо из экспериментальной кривой титрования (рис. 1).
Для исследования образования гетеробиядерных комплексов на основе глутатионатных комплексов золота(I) были выбраны ионы Cu2+ и Zn2+. Хорошо известно, что медь(II) образует высокоустойчивые пятичленные циклы с ГФ обычных α-аминокислот. Так, для глицина равновесия Cu2+ + iGly– ↔ ${\text{Cu(Gly}})_{i}^{{2 - i}}$ имеют lgβ1 = 8.2 и lgβ2 = 15.1 [20]. Уже при небольшом избытке глицина влияние гидроксидных комплексов незначительно. Устойчивость глицинатных комплексов цинка(II) намного ниже (lgβ1 = 4.8), и, в отличие от меди(II), большее значение имеет равновесие Zn2+ + Gly– + OH– ↔ Zn(Gly)OH0 с lgβOH = 10.7 [20], т.е. при небольшом избытке глицина в слабощелочной области основными формами являются Zn(Gly)+ и Zn(Gly)OH.
В состав глутатиона также входят две пептидные –NH-группы. Известно, что медь(II) способна образовывать комплексы с координацией к депротонированной группе (–N––) [21–24]. При не очень высоких значениях pH обычно образуется несколько пятичленных (реже шестичленных) циклов, причем в качестве одной из групп часто выступает –NH2. Однако при расположении глутаминовой кислоты, как в глутатионе (γ-Glu–Cys–Gly), хелатный цикл (NH2, N–) может быть только семичленным, т.е. крайне неустойчивым. Тем не менее для более надежных выводов требуются данные для аналогичных систем. При этом прямое комплексообразование меди(II) с глутатионом невозможно вследствие восстановления меди(II) до меди(I). Поэтому в качестве модельной была выбрана система с дисульфидом глутатиона (GSSG), в котором группа –S–S– не окисляется медью(II) и не участвует в комплексообразовании, но при этом в молекуле GSSG полностью сохраняется строение остальной части глутатиона. В работе [25] было показано, что при умеренных значениях рН (7.4) в водном растворе при комплексообразовании Cu2+ с GSSG реализуется только обычный способ координации меди(II) с ГФ остатка глутаминовой кислоты без участия пептидных групп. Аналогичные выводы сделаны в работе [26].
В ходе наших экспериментов с медью(II) к растворам с CGS : CAu = 1–2 с pH 8.0–8.5 добавляли раствор CuCl2. Однако оказалось, что при CGS : CAu > 1.5 после добавления CuCl2 раствор сразу становился коричневым, а затем через 10–20 с полностью обесцвечивался. Очевидно, это связано с окислением части глутатиона медью(II) с переходом ее в медь(I). Для уменьшения влияния этого процесса требовалось снижение [GS*]. Лишь при CGS : CAu ≤ 1.5 (CAu = 1 × 10–3 моль/л) признаков редокс-процесса не наблюдалось и получаемые растворы имели чистый голубой цвет.
К растворам с выбранными значениями CGS : : CAu из диапазона 1–1.5 и pH ~ 8, при котором n* ~ ~ 0.95, добавляли CuCl2 небольшими (CCu < CAu) порциями с одновременным точным измерением pH. После добавления каждой порции наблюдали значительное (до трех единиц) снижение pH раствора. Его “восстанавливали” до приблизительно исходного значения добавлением щелочи, а затем добавляли следующую порцию CuCl2. После добавления нескольких порций CuCl2 величина pH раствора больше не изменялась. Результаты двух серий (из пяти) для CAu = 1.0 × 10–3 и 1.0 × 10–2 моль/л показаны на рис. 2. Их можно объяснить следующим образом. Снижение pH раствора после добавления порции CuCl2 однозначно свидетельствует о взаимодействии Cu2+ с протонированными группами $--{\text{NH}}_{3}^{ + },$ приводящем к выделению H+. Поскольку каждый глицинатный фрагмент содержит не более одного иона H+, по количеству выделившихся ионов H+ можно определить, сколько ГФ оказались связанными с одним ионом Cu2+. Уже из количества щелочи (ΔnOH), затрачиваемой на “восстановление” pH после добавления порций CuCl2 (ΔnCu), следовало, что количество ионов H+, выделяемых на каждый ион Cu2+ (k), составляет ~2 (kCu = ΔnOH/ΔnCu ~ 2). Об этом же свидетельствует тот факт, что pH раствора перестает значительно изменяться при добавлении CuCl2, когда общее количество добавленной меди(II) достигает ~1/2 CGS. Более точный расчет проводили по уравнению материального баланса для H+ с пренебрежением слагаемыми [H+] и [OH–]:
(3)
$C_{{\text{H}}}^{*} = n*\left( {{{C}_{{{\text{GS}}}}}--{{k}_{{{\text{Cu}}}}}{{C}_{{{\text{Cu}}}}}} \right),$Рис. 2.
Изменение pH растворов при добавлении CuCl2 порциями. Вертикальные стрелки – добавление CuCl2, наклонные стрелки – добавление NaOH для “восстановления” pH. 1 – CAu = 1.0 × 10–3 моль/л, GS : Au = 1.5 : 1, ΔnCu = 0.0098 ммоль; 2 – CAu = 1.0 × × 10–2 моль/л, GS : Au = 1 : 1, ΔnCu = 0.049 ммоль. V = = 40 мл.
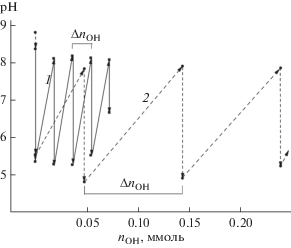
Рис. 3.
Сравнение спектров. 1 – (AuGS)m${\text{Cu}}_{{xm}}^{*}$ (раствор GS : Au : Cu = 1 : 1 : 0.4, CAu = 1.0 × 10–2 моль/л, l = 5 см, ${\text{(AuGS)}}_{m}^{*}$ в видимой области не поглощает, xср = 0.4); 2 – Cu(Gly)2 (раствор Gly : Cu = 2.1 : 1, CCu = = 1.0 × 10–2 моль/л, l = 1 см); 3 – раствор Gly : Cu = 1 : 1, CCu = 1.0 × 10–2 моль/л, l = 1 см. ε* = A/lCCu, I = 0.2 M (NaCl), pH 8.0.
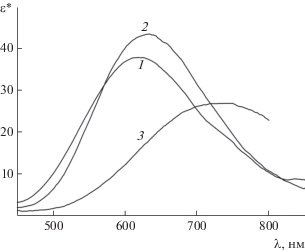
На наш взгляд, факт связывания одного иона Cu2+ с двумя ГФ является наиболее интересной особенностью гетеробиядерных комплексов с медью(II) в растворе. Причем это наблюдается как для растворов, в которых присутствует только ${\text{(AuGS)}}_{m}^{*}$ (CGS : CAu = 1), так и для растворов, содержащих одновременно ${\text{(AuGS)}}_{m}^{*}$ и ${\text{Au(GS)}}_{2}^{*}.$ Поскольку в состав глутатиона входит только один ГФ, ион Cu2+ координирует ГФ двух глутатионатов. Остается неясным, принадлежат ли эти глутатионат-ионы двум разным молекулам комплексов или входят в состав одной молекулы комплекса, полимерного ${\text{(AuGS)}}_{m}^{*}$ или мономерного ${\text{Au(GS)}}_{2}^{*}.$ По аналогии с дисульфидными комплексами Cu(GSSG)* [25, 26] в последнем случае возможный способ координации Cu2+ в (AuGS)m${\text{Cu}}_{{xm}}^{*}$ (x < 0.5) можно представить в виде
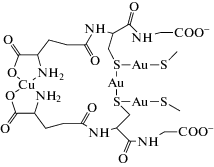
Фрагмент –S(Au)–Au–S(Au)– в случае Cu(GSSG)* представляет собой просто –S–S–, а для ${\text{Au(GS)}}_{2}^{*}$ – это –S–Au–S–. Хотя схематично процесс образования комплекса можно представить уравнением:
(4)
$\begin{gathered} {\text{C}}{{{\text{u}}}^{{2 + }}} + 2{\text{NH}}_{3}^{ + }{\kern 1pt} --{\kern 1pt} {\text{CH}}\left( {\text{R}} \right){\kern 1pt} --{\kern 1pt} {\text{CO}}{{{\text{O}}}^{--}} \leftrightarrow \\ \leftrightarrow {\text{Cu}}{{({\text{N}}{{{\text{H}}}_{2}}{\kern 1pt} --{\kern 1pt} {\text{CH}}\left( {\text{R}} \right){\kern 1pt} --{\kern 1pt} {\text{CO}}{{{\text{O}}}^{--}})}_{2}} + 2{{{\text{H}}}^{ + }}, \\ \end{gathered} $При дальнейшем увеличении CCu : CGS > 0.5 наблюдается выделение голубого осадка. Величина kCu при этом снижается. Осадок, полученный при исходном соотношении Au : GS : Cu = 1 : 1 : 1 и pH 6.0, имеет чистый голубой цвет. После фильтрования (фильтруется плохо) и промывки водой и этанолом его сушили на воздухе. Результаты элементного анализа на Au, Cu, C, H, N, S показали, что их содержание отвечает формуле AuCu(GSH)⋅0.5SO4⋅1.5H2O. Сульфат-ион, компенсирующий часть заряда Cu2+, изначально присутствует в растворе вследствие окисления сульфита при получении ${\text{AuCl}}_{2}^{ - }$ из ${\text{AuCl}}_{4}^{ - }.$ В ИК-спектре осадка, в отличие от спектра GSH3 (рис. S1 ), отсутствует полоса при 2525 см–1, отвечающая –SH, что характерно для полимерных комплексов с координацией иона металла к тиольной группе [27], но присутствуют полосы при 881 и 431 см–1, которые можно отнести к колебаниям Cu–${\text{SO}}_{4}^{{2 - }}$ и Cu–N [28, 29].
Аналогичные эксперименты были выполнены с цинком(II). По сравнению с медью(II) (рис. 2) при добавлении ZnCl2 порциями к растворам с CGS : CAu = 1–2 изменения pH были намного меньше: ~1 и ниже (см. Приложение, рис. S2 ). Расчеты, аналогичные описанным выше, показывают, что каждый ион Zn2+ связывается не более чем с одним ГФ глутатиона, координированного к золоту(I). По достижении соотношения CZn : CGS ~ 0.5 наблюдается появление белого осадка. Очевидная причина состоит в том, что комплексы цинка(II) с лигандами, содержащими ГФ, намного менее устойчивы, чем комплексы меди(II).
Нас также интересовала возможность флуоресценции гетеробиядерного комплекса с цинком(II). Раствор исходного комплекса ${\text{(AuGS)}}_{m}^{*}$ сам проявляет слабую флуоресценцию (λmax = 430 и 650 нм), однако введение Zn2+ до CZn : CGS = 0.4 не добавило новых особенностей (см. Приложение, рис. S3 ).
Аналогично глутатиону ГФ присутствует в цистеине ((${\text{NH}}_{3}^{ + }$)–CH(CH2–SH)–COO–). Однако полимер [Au(HCys)]m чрезвычайно плохо растворим. Его растворение происходит лишь при pH > > 7–8 и при значительном (>2.2/1) избытке цистеина. В этих условиях использование меди(II) невозможно, поскольку она легко окисляет цистеин и переходит в медь(I). Эксперименты с цинком(II) также оказались неудачными из-за образования белых осадков во всей интересующей нас области (pH 7–9.5, CCys : CAu = 2.2–2.5, CZn : CAu = 0.2–1).
ЗАКЛЮЧЕНИЕ
Глутатионатные комплексы золота(I) (как полимерные ${\text{(AuGS)}}_{m}^{*},$ так и мономерные ${\text{Au(GS)}}_{2}^{*}$) образуют в водном растворе гетеробиядерные комплексы с ионами Cu2+ и Zn2+, которые связываются с ГФ глутатионата, координированного к золоту(I) через атом S. В случае меди(II) один ион Cu2+ связан с двумя глутатионатами. Для цинка(II) один ион Zn2+ связан не более чем с одним глутатионатом. При pH 6–9, когда карбоксильные группы глутатиона депротонированы, для полимерного комплекса наиболее вероятный состав гетеробиядерных комплексов в растворе отвечает формулам (AuGS)mCumx${\text{H}}_{{m - 2mx}}^{{m - }}$ и (AuGS)m(ZnOH)mx${\text{H}}_{{m - mx}}^{{m - }}$ с переменным количеством M2+ (x < 0.5). Во всех случаях при CM2+ : CGS > > 0.5 гетеробиядерные комплексы выделяются в виде твердой фазы. В отличие от раствора, для меди(II) соотношение основных компонентов Au : GS : Cu в ней соответствует 1 : 1 : 1.
Список литературы
Макотченко Е.В., Байдина И.А., Корольков И.В. // Журн. неорган. химии. 2019. Т. 64. № 11. С. 23. [Makotchenko E.V., Baidina I.A., Korol’kov I.V. // Russ. J. Inorg. Chem. 2019. V. 64. № 11. P. 41. https://doi.org/10.1134/S0036023619 010157]
Черкашина Н.В., Чураков А.В., Якушев И.А. и др. // Коорд. химия. 2019. Т. 45. № 4. С. 197. [Cherkashina N.V., Churakov A.V., Yakushev I.A. et al. // J. Coord. Chem. 2019. V. 45. № 4. Р. 253. https://doi.org/10.1134/S107032841904002X]
Жигулин Г.Ю., Забродина Г.С., Каткова М.А., Кетков С.Ю. // Коорд. химия. 2019. Т. 45. № 5. С. 306. [Zhigulin G.Y., Zabrodina G.S., Katkova M.A., Ketkov S.Y. // J. Coord. Chem. 2019. V. 45. № 5. Р. 356. https://doi.org/10.1134/S107032841905004X]
Wenzel M., Bigaeva E., Richard P. et al. // J. Inorg. Biochem. 2014. V. 141. P. 10. https://doi.org/10.1016/j.jinorgbio.2014.07.011
Wenzel M., de Almeida A., Bigaeva E. et al. // Inorg. Chem. 2016. V. 55. P. 2544. https://doi.org/10.1021/acs.inorgchem.5b02910
Majzik A., Fülöp L., Csapó E. et al. // Colloids Surf. B. 2010. V. 81. P. 235. https://doi.org/10.1016/j.colsurfb.2010.07.011
Bieri M., Bürgi T. // Phys. Chem. Chem. Phys. 2006. V. 8. P. 513. https://doi.org/10.1039/b511146c
Ao H., Feng H., Li K. et al. // Sens. Actuators, B: Chem. 2018. V. 272. P. 1. https://doi.org/10.1016/j.snb.2018.05.151
Söptei B., Mihály J., Szigyártó I.Cs. et al. // Colloids Surf., A: Physicochem. Eng. Asp. 2015. V. 470. P. 8. https://doi.org/10.1016/j.colsurfa.2015.01.048
Luo Z., Yuan X., Yu Y. et al. // J. Am. Chem. Soc. 2012. V. 134. P. 16662. https://doi.org/10.1021/ja306199p
Brown D.H., Smith W.E. // J. Chem. Soc., Dalton Trans. 1980. V. 9. P. 217.
Shaw III C.F. // Chem. Rev. 1999. V. 99. P. 2589. https://doi.org/10.1021/cr980431o
Mohr F. Gold Chemistry. Weinheim: Wiley-VCH Verlag GmbH & Co., 2009.
Mironov I.V., Kharlamova V.Yu. // J. Solution Chem. 2018. V. 47. P. 511. https://doi.org/10.1007/s10953-018-0735-y
Gammons C.H., Yunmei Y., Wiliams-Jones A.E. // Geochim. Cosmochim. Acta. 1997. V. 61. P. 1971. https://doi.org/10.1016/S0016-7037(97)00060-4
Миронов И.В., Харламова В.Ю. // Журн. неорган. химии. 2016. Т. 61. № 1. С. 129. [Mironov I.V., Kharlamova V.Yu. // Russ. J. Inorg. Chem. 2016. V. 61. № 1. P. 123. https://doi.org/10.1134/S0036023616010174]
Harned H.S., Owen B.B. The Physical Chemistry of Electrolytic Solutions. N.Y.: Reinhold, 1950.
Mironov I.V., Kharlamova V.Yu. // J. Solution Chem. 2020. V. 49. P. 583. https://doi.org/10.1007/s10953-020-00994-0
Nagy P., Winterbourn C.C. // Adv. Mol. Toxicol. 2010. V. 4. P. 189. https://doi.org/10.1016/S1872-0854(10)04006-3
Kiss T., Sovago I., Gergely A. // Pure & Appl. Chem. 1991. V. 63. № 4. P. 597. https://doi.org/10.1351/pac199163040597
Usacheva T.R., Pham Thi L., Kuzmina K.I., Sharnin V.A. // J. Therm. Anal. Calorim. 2017. V. 130. P. 471. https://doi.org/10.1007/s10973-017-6207-6
Kozlowski H., Bal W., Dyba M., Kowalik-Jankowska T. // Coord. Chem. Rev. 1999. V. 184. P. 319. https://doi.org/10.1016/S0010-8545(98)00261-6
Miyoshi K., Sugiura Y., Ishizu K. et al. // J. Am. Chem. Soc. 1980. V. 102. № 19. P. 6131. https://doi.org/10.1021/ja00539a027
Sóvágó I., Kállay C., Várnagy K. // Coord. Chem. Rev. 2012. V. 256. P. 2225. https://doi.org/10.1016/j.ccr.2012.02.026
Pedersen J.Z., Steinkühler C., Weser U., Rotilio G. // Biometals. 1996. V. 9. № 1. P. 3. https://doi.org/10.1007/BF00188083
Sóvágó I., Katalin O. // Dalton Trans. 2006. P. 3841. https://doi.org/10.1039/b607515k
Farrag M., Mohamed R.A. // J. Photochem. Photobiol., A: Chem. 2016. V. 330. P. 117. https://doi.org/10.1016/j.jphotochem.2016.07.027
Berger J. // J. Raman Spectrosc. 1976. V. 5. P. 103. https://doi.org/10.1002/jrs.1250050202
Herlinger A.W., Wenhold S.L., Veach Long II T. // J. Am. Chem. Soc. 1970. V. 92. P. 6474. https://doi.org/10.1021/ja00725a015
Дополнительные материалы
- скачать ESM.docx
- Рис. S1. ИК спектры.
Рис. S2. Изменение pH растворов при добавлении ZnCl2 порциями.
Рис. S3. Спектры флуоресценции растворов.
Инструменты
Журнал неорганической химии