

Журнал неорганической химии, 2021, T. 66, № 9, стр. 1192-1246
НАПРАВЛЯЕМАЯ ФУНКЦИОНАЛЬНЫМИ ГРУППАМИ B–H-АКТИВАЦИЯ ПОЛИЭДРИЧЕСКИХ ГИДРИДОВ БОРА КОМПЛЕКСАМИ ПЕРЕХОДНЫХ МЕТАЛЛОВ (ОБЗОР)
И. Б. Сиваев *
Институт элементоорганических соединений им. А.Н. Несмеянова РАН
119991 Москва, ул. Вавилова, 28, Россия
* E-mail: sivaev@ineos.ac.ru
Поступила в редакцию 22.04.2021
После доработки 27.04.2021
Принята к публикации 29.04.2021
Аннотация
Активация В–Н-групп полиэдрических гидридов бора (боранов и карборанов) является одним из наиболее активно развивающихся направлений химии бора и представляет значительный интерес для специалистов в области органической химии, металлоорганического катализа и координационной химии. В данном обзоре обобщены сведения по направляемым функциональными группами B–H реакциям активации полиэдрических гидридов бора комплексами переходных металлов, начиная с ранних работ 1980-х гг. и заканчивая самыми последними достижениями в этой области. Рассмотрено образование комплексов переходных металлов со связями M…H–B и M–B и их использование в синтезе различных органических производных (кар)боранов.
ВВЕДЕНИЕ
За более чем полвека, прошедших со времени открытия полиэдрических боранов и карборанов, они продемонстрировали высокий потенциал использования в самых различных областях, начиная с создания новых материалов и заканчивая медициной [1–5]. Это во многом обусловлено интенсивным развитием их химии и разработкой методов синтеза их различных производных [3, 6–11]. При этом еще несколько лет назад казалось, что потенциал развития методов функционализации этих систем уже практически исчерпан, а химикам, работающим в этой области, остается решать лишь рутинные задачи или, наоборот, ставить перед собой весьма экзотические цели, такие как создание наномашин [12, 13] или молекулярных переключателей [14–17] на основе карборанов.
Революционный прорыв в этой области случился в 2014 г., когда было предложено использовать заранее введенные в карборан первичные функциональные группы в качестве направляющих лигандов для введения различных вторичных заместителей через катализируемую переходными металлами B–H-активацию карборанового остова [18]. Это существенно упростило введение заместителей в те положения, которые были недоступны прямому замещению (положения 3 и 6), а также сделало возможным введение различных заместителей в те положения, которые раньше были недоступны замещению вообще (например, положения 4 и 5). Это направление получило быстрое развитие. Уже в 2019 г. был опубликована первая обзорная статья [19], а количество публикаций, посвященных данной тематике, продолжает увеличиваться с каждым годом. При этом необходимо отметить, что реакции B–H-активации карборанового остова с использованием заместителей в качестве вспомогательных лигандов, приводящие к образованию циклометаллированных комплексов со связью бор–металл, впервые были описаны еще в середине 70-х годов прошлого века [20] и затем активно исследовались в 1980-х годах [21]. Однако в силу разных причин эти два взаимосвязанных процесса изучались раздельно разными исследовательскими группами, одни из которых концентрировались на синтезе и изучении самих B-металлированных комплексов, а другие – на разработке методов синтеза новых производных карборанов и боранов, в которых такие комплексы играют роль промежуточных продуктов каталитического цикла.
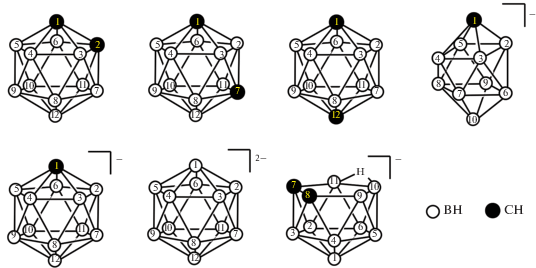
В данном обзоре предпринята попытка дать максимально полную картину реакций B–H-активации полиэдрических боранов и карборанов комплексами переходных металлов с использованием заместителей, содержащих различные донорные атомы, в качестве вспомогательных лигандов, включая как образование комплексов со связями B–H…M и B–M, так и их использование в синтезе разнообразных производных боранов и карборанов. В качестве объектов B–H-активации рассматриваются в первую очередь нейтральные икосаэдрические карбораны C2B10H12, а также анионные борные кластеры 7,8-дикарба-нидо-ундекаборат (нидо-карборан) [7,8-C2B9H12]–, 1-карба-клозо-декаборат [1-CB9H10]–, карба-клозо-додекаборат [CB11H12]– и клозо-додекаборат [B12H12]2–-анионы (рис. 1).
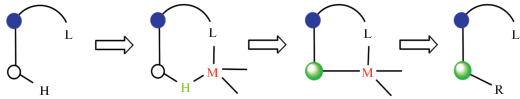
Как и в случае катализируемых переходными металлами реакций внутримолекулярной C–H-активации [22–26], предполагемый механизм реакций B–H-активации включает несколько стадий, первой из которых является координация переходного металла донорным атомом первичного заместителя-лиганда с последующим замещением одного из лабильных лигандов гидридом BH, находящимся в орто-положении к замещенному атому. Затем происходит окислительное внедрение металла в связь B–H, восстановительное отщепление гидрида (как правило, в виде кислоты HX) с последующей активацией связи C–X органического реагента (вторичного заместителя) и внедрением вторичного заместителя в связь B–M с образованием более устойчивой связи B–C (рис. 2). При этом устойчивость образующихся промежуточных комплексов зависит от целого ряда факторов, как электронных (включая природу переходного металла и его лигандного окружения, донорные свойства первичного заместителя, степень гидридности связи B–H), так и стерических (удаленность донорного атома заместителя от (кар)боранового остова, наличие вблизи реакционного центра других заместителей и т.п.).
Рис. 2.
Общая схема направляемой функциональными группами B–H-активации полиэдрических гидридов бора с участием комплексов переходных металлов.
В данном обзоре реакции B–H-активации полиэдрических гидридов бора рассмотрены в порядке уменьшения донорных свойств первичного заместителя в ряду P(As) > N > O(S). Это дает возможность, с одной стороны, рассмотреть их в историческом развитии, а с другой – провести черту между стабильными и более реакционноспособными циклометаллированными комплексами, которые активно используются для введения в борный остов органических заместителей.
Реакции B–H-активации с участием фосфинов как направляющих лигандов
Благодаря хорошей синтетической доступности разнообразных карборанил-фосфиновых лигандов [26] к настоящему времени получено огромное количество комплексов переходных металлов на их основе [27–30]. Следует отметить, что присоединение фосфина к атому углерода карборанового остова приводит к значительному понижению его донорного характера вследствие сильного электроноакцепторного влияния C-карборанильной группы, которое может быть частично “погашено” внедрением метиленового спейсера между атомом фосфора и карборановым остовом. С точки зрения направляющего действия фосфинового лиганда, присоединенного к атому углерода C(1) орто-карборана, доступными для B–H-активации являются атомы бора в положениях B(3) и B(6), а также B(4) и B(5) (рис. 1). При этом первые из них (в положениях B(3) и B(6)) характеризуются меньшей электронной плотностью, что способствует образованию связи бор–металл, однако наличие дополнительного заместителя в соседнем положении C(2) может затруднить атаку металла и сделать более предпочтительной B–H-активацию по положениям B(4) и B(5).
Как правило, C,C'-дифосфиновые лиганды на основе орто-карборана образуют P,P'-хелатные комплексы, в то время как для C-монофосфиновых лигандов характерно образование P,B-хелатных комплексов с активацией связи B–H. Так, кипячение комплекса транс-[PdCl2(1-Ph2PCH2-1,2-C2B10H11)2] в толуоле приводит к элиминированию HCl с образованием смеси содержащих пятичленные палладоциклы мономерного [PdCl(1-Ph2PCH2-1,2-C2B10H10-κ2-P,B(3/4))(1-Ph2PCH2-1,2-C2B10H11-κ1-P)] и димерного [PdCl(1-Ph2PCH2-1,2-C2B10H10-κ2-P,B(3/4))]2 комплексов, которые могут быть разделены кристаллизацией из толуола–гексана [31, 32]. Аналогичным образом кипячение карбораниларсинового комплекса транс-[PdCl2(1-Me2AsCH2-1,2-C2B10H11)2] в толуоле приводит к смеси [PdCl(1-Me2AsCH2-1,2-C2B10H10-κ2-As,B(3/4))(1-Me2AsCH2-1,2-C2B10H11-κ1-As)] и [PdCl(1-Me2AsCH2-1,2-C2B10H10-κ2-As,B(3/4))]2 [32]. В то же время при кипячении транс-[PdCl2(1-Ph2PCH2-2-R-1,2-C2B10H10)2] (R = = H, Me) в изопропаноле образуются исключительно димерные комплексы [PdCl(1-Ph2PCH2-2-R-1,2-C2B10H9-κ2-P,B(3/4))]2 [32]. Последние также могут быть получены обменными реакциями циклопалладированного N,N-дибензиламино комплекса [PdCl(2-Me2NCH2C6H-κ2-N,C)]2 с соответствующими карборанилфосфиновыми лигандами в кипящем хлороформе [32, 33]. Димерные комплексы [PdX(1-Ph2PCH2-1,2-C2B10H10-κ2-P,B(3/4))]2 (X = Cl, Br, I) были получены реакцией [PdCl(η3-Allyl)]2 с 1-Ph2PCH2-1,2-C2B10H11 в уксусной кислоте в присутствии соответствующих галогенидов лития [32]. При обработке пиридином они легко разрываются с образованием соответствующих мономерных комплексов [(Py)PdX(1-Ph2PCH2-1,2-C2B10H10-κ2-P,B(3/4))]2 [31, 32]. Следует отметить, что, согласно данным ЯМР-спектроскопии, все эти реакции приводят к смеси продуктов B(3)- и B(4)-активации [32]. Аналогичным образом кипячение комплекса платины транс-[PtCl2(1-Ph2PCH2-1,2-C2B10H11)2] в толуоле приводит к циклоплатинированному комплексу [PtCl(1-Ph2PCH2-1,2-C2B10H10-κ2-P,B))(1-Ph2PCH2-1,2-C2B10H11-κ1-P)] [31].
Взаимодействие карборанилфосфинов 1-R2P-2-R'-1,2-C2B10H10 (R = t-Bu, Ph; R' = H, Me, i-Pr) с Na2[PdCl4] в метаноле или [(PhCN)2PdCl2] в бензоле или тетрагидрофуране приводит к содержащим четырехчленные палладоциклы мономерным [PdCl(1-Ph2P-1,2-C2B10H10-κ2-P,B)(1-Ph2P-1,2-C2B10H11-κ1-P)] и димерным [PdCl(1-R2P-2-R'-1,2-C2B10H9-κ2-P,B)]2 (R = t-Bu, R' = H; R = Ph, R' = H, Me, i-Pr) комплексам (схема 1 ) [32, 34]. Димерные комплексы [PdX(1-Ph2P-2-R-1,2-C2B10H9-κ2-P,B)]2 (R = H, Me, i-Pr) также были получены обменными реакциями циклопалладированного N,N-дибензиламино комплекса [PdCl(2-Me2NCH2C6H-κ2-N,C)]2 с соответствующими карборанилфосфиновыми лигандами в кипящем хлороформе [32, 33]. При этом, согласно данным ЯМР-спектроскопии, реакции c 1-Ph2P-1,2-C2B10H11 и 1-Ph2P-2-Me-1,2-C2B10H10 приводят к смеси продуктов B(3)- и B(4)-активации, тогда как реакции с содержащим при втором атоме углерода карборана объемный заместитель 1-Ph2P-2-i-Pr-1,2-C2B10H10 дают исключительно продукт B(4)-активации [32]. В свою очередь, в случае 1-t-Bu2P-1,2-C2B10H11 реакция приводит к селективному образованию продукта B(3)-активации [PdCl(1-t-Bu2P-1,2-C2B10H10-κ2-P,B(3))]2 (рис. 3).
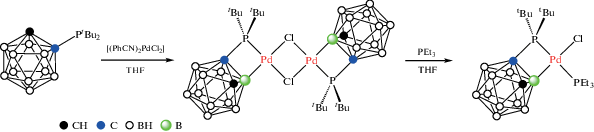
Схема 1.
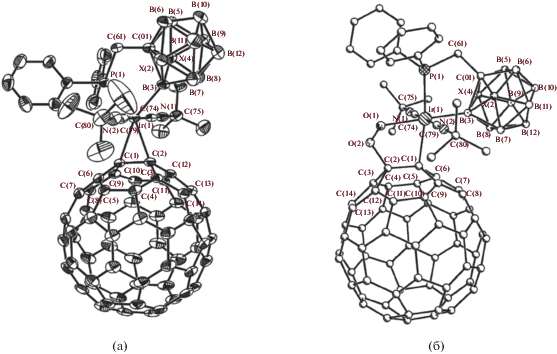
Рис. 3.
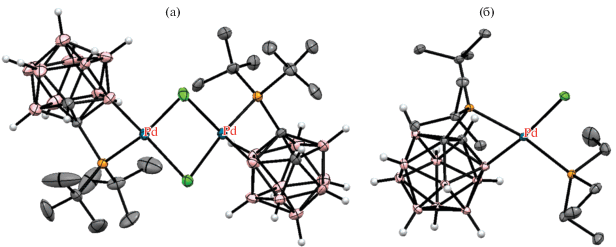
Строение комплексов [PdCl(1-t-Bu2P-1,2-C2B10H10-κ2-P,B(3))]2 (а) и транс-[(Et3P)PdCl(1-t-Bu2P-1,2-C2B10H10-κ2-P,B(3))] (б). Атомы водорода органических заместителей и лигандов не показаны для ясности.
Димерный комплекс [PdCl(1-t-Bu2P-1,2-C2B10H10-κ2-P,B(3))]2 легко разрывается при обработке триэтилфосфином в тетрагидрофуране с образованием транс-[(Et3P)PdCl(1-t-Bu2P-1,2-C2B10H10-κ2-P,B(3))] (рис. 3, схема 1 ) [34]. В то же время реакция [PdCl(1-Ph2P-2-i-Pr-1,2-C2B10H9-κ2-P,B(4))]2 с 1‑Ph2PCH2-1,2-C2B10H11 в смеси хлороформа и уксусной кислоты приводит к замене четырехчленного палладацикла более устойчивым пятичленным [PdCl(1-Ph2PCH2-1,2-C2B10H10-κ2-P,B)]2 [32, 35].
Следует отметить, что реакция 1-дифенилфосфино-орто-карборана с арилгалогенидами в присутствии Li2CO3 и каталитических количеств PdCl2 приводит к B–H-активации с образованием соответствующих 1-дифенилфосфино-3-галоген-орто-карборанов 1-Ph2P-3-X-1,2-C2B10H10 (X = Cl, Br, I), в то время как при использовании более сильного основания (t-BuOLi) в реакциях с арилбромидами образуются продукты C-арилирования 1-Ph2P-2-Ar1,2-C2B10H10 (схема 2 ) [36]:
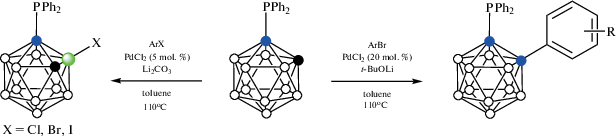
Схема 2.
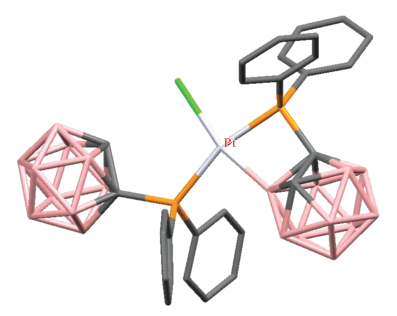
Комплекс с четырехчленным платинациклом [PtCl(1-Ph2P-1,2-C2B10H10-κ2-P,B(3))(1-Ph2P-1,2-C2B10H11-κ1-P)] (рис. 4) был получен при перекристаллизации хелатного комплекса [PtCl2(1-PPh2-2-PF2-1,2-C2B10H10-κ2-P,P')] [37].
Рис. 4.
Строение комплекса [PtCl(1-Ph2P-1,2-C2B10H10-κ2-P,B(3))(1-Ph2P-1,2-C2B10H11-κ1-P)] (атомы водорода не показаны для ясности).
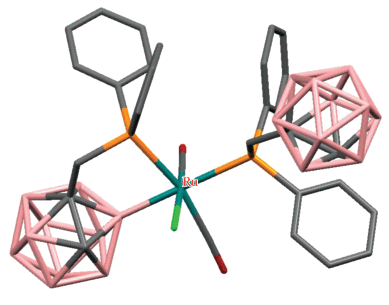
Циклометаллированные карборанилфосфиновые комплексы родия и иридия были получены на основе 1-дифенилфосфинометил-орто-карборана. Взаимодействие 1-Ph2PCH2-1,2-C2B10H11 с комплексами родия(I) [(CO)2RhCl]2, [(C8H14)2RhCl]2 или [(COD)RhCl]2 в кипящем гексане в присутствии пиридина или его производных приводит к соответствующим B(3)-металлированным комплексам родия(III) [RhHCl(1-Ph2PCH2-1,2-C2B10H10-κ2-P,B(3))(NC5H4R)2] (R = H, 3-Me, 4-Me, 4-OMe) (схема 3). Предполагается, что реакция протекает через образование комплекса транс-[(CO)RhCl(1-Ph2PCH2-1,2-C2B10H11-κ1-P)2], который был выделен при проведении реакции при комнатной температуре в отсутствие пиридина [38, 39]. Аналогичный иодидный комплекс [RhHI(1-Ph2PCH2-1,2-C2B10H10-κ2-P,B(3))(NC5H4-4-Me)2] был получен исходя из [(COD)RhI]2 [38]. Реакции 1-Ph2PCH2-1,2-C2B10H11 с [(COD)RhCl]2 приводят к соответствующим комплексам родия(III) и иридия(III) [(COD)MHCl(1-Ph2PCH2-1,2-C2B10H10-κ2-P,B(3))], где M = Rh, Ir (схема 3 ) [38, 39]. Комплексы [(Py)2RhHCl(1-Ме2AsCH2-1,2-C2B10H10-κ2-P,B(3))] и [(COD)IrHCl(1-Me2AsCH2-1,2-C2B10H10-κ2-P,B(3))] были получены аналогичным образом исходя из 1-диметиларсинометил-орто-карборана [39].
Реакция 1-Ph2PCH2-1,2-C2B10H11 с моногидридным комплексом иридия(I) [(Ph3P)3IrH(CO)] приводит к окислительному B(3)-циклометаллированию с образованием смеси диастереомерных дигидридных комплексов иридия(III) [(Ph3P)(CO)IrH2(1-Ph2PCH2-1,2-C2B10H10-κ2-P,B(3))]. При избытке карборанилфосфина происходит двойное циклометаллирование с образованием моногидридного комплекса [(CO)IrH(1-Ph2PCH2-1,2-C2B10H10-κ2-P,B(3))2] (рис. 5) в виде смеси диастереомеров [40]. Реакция 1-Ph2PCH2-1,2-C2B10H11 с моногидридным комплексом иридия(I) [(COD)(Ph3P)IrH] в гексане приводит к B(3)-циклометаллированию с образованием дигидридного комплекса иридия(III) [(Ph3P)2IrH2(1-Ph2PCH2-1,2-C2B10H10-κ2-P,B(3))] (рис. 5) [40].
Рис. 5.
Строение комплексов [(CO)IrH(1-Ph2PCH2-1,2-C2B10H10-κ2-P,B(3))2] (а) и [(Ph3P)2IrH2(1-Ph2PCH2-1,2-C2B10H10-κ2-P,B(3))] (б). Атомы водорода органических заместителей и фенильные группы лигандов не показаны для ясности.
Взаимодействие циклометаллированных дигидридных комплексов [IrH2(1-Ph2PCH2-1,2-C2B10H10-κ2-P,B)(PPh3)(L)] (L = PPh3, CO) с фуллереном C60 в присутствии t-BuNC в кипящем толуоле приводит к элиминированию водорода с образованием комплекса [(η2-C60)Ir(1-Ph2PCH2-1,2-C2B10H10-κ2-P,B(3))(t-BuNC)2], содержащего в координационной сфере металла карборанилфосфиновый и фуллереновый лиганды (рис. 6) [41]. Окисление последнего кислородом в хлороформе приводит к избирательному внедрению молекулы кислорода в связь Ir–C в транс-положении к атому бора карборана с образованием комплекса [σ-C60OO-Ir(1-Ph2PCH2-1,2-C2B10H10-κ2-P,B)(t-BuNC)2] [42].
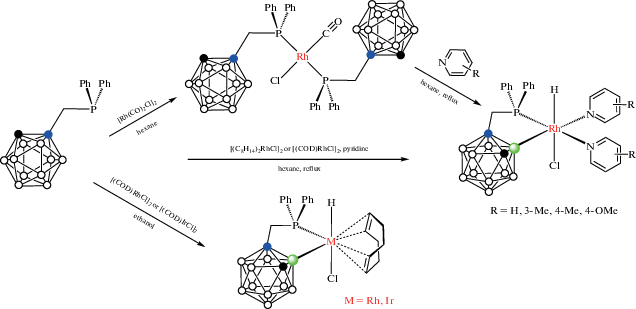
Схема 3.
Реакция 1-диметилфосфино-орто-карборана 1-Me2P-1,2-C2B10H11 с комплексом иридия(I) [Ir(C8H14)2IrCl]2 в кипящем циклогексане приводит к окислительному внедрению металла в связь B(3)–H с образованием комплекса иридия(III) [IrHCl(1-Me2P-1,2-C2B10H11-κ1-P)2(1-Me2P-1,2-C2B10H10-κ2-P,B(3))] (схема 4 ) [20]. Реакция 1-диизопропилфосфино-орто-карборана 1-i-Pr2P-1,2-C2B10H11 с [(COD)IrCl]2 в гексане при комнатной температуре приводит к циклометаллированному комплексу [(COD)IrHCl(1-i-Pr2P-1,2-C2B10H10-κ2-P,B(3))] (схема 4 , рис. 7) [43].
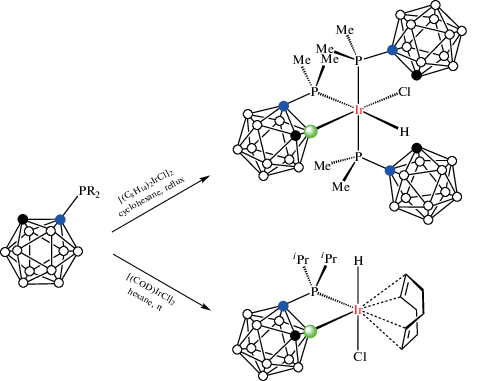
Схема 4.
Рис. 7.
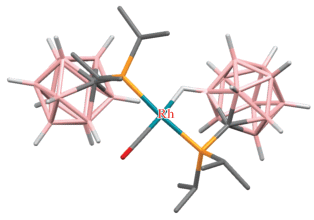
Строение комплексов [(COD)IrHCl(1-i-Pr2P-1,2-C2B10H10-κ2-P,B(3))] (а) и [Rh(μ-Cl)(CO)(1-t-Bu2P-1,2-C2B10H10-κ2-P,B(3))]2 (б). Атомы водорода органических заместителей и лигандов не показаны для ясности.
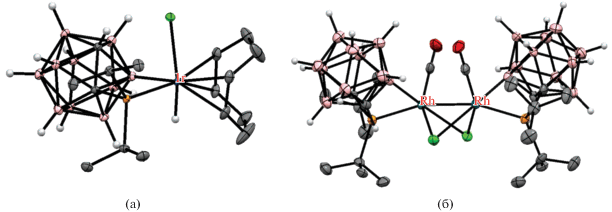
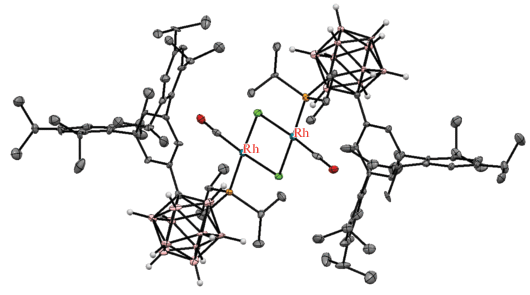
Реакция 1-ди-трет-бутилфосфино-орто-карборана с комплексом родия(I) [(CO)2RhCl]2 в толуоле неожиданно привела к образованию циклометаллированного диродиевого(III) комплекса [Rh(μ-Cl)(CO)(1-t-Bu2P-1,2-C2B10H10-κ2-P,B(3))]2 (схема 5 , рис. 7) [32].
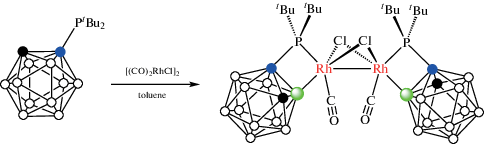
Схема 5.
Образование циклометаллированных комплексов является ключевой стадией Rh-катализируемого 3-моно- и 3,6-диарилирования 1-дифенилфосфино-орто-карборана арилбромидами, а также 3,6-диалкилирования винил- и аллилбензолами (схема 6 ) [36].
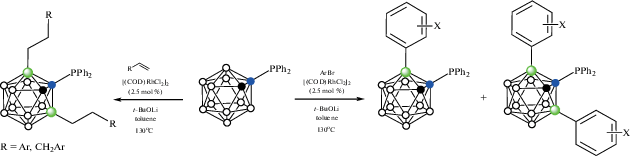
Схема 6.
Показано, что B–H-активация может быть заблокирована введением объемных заместителей при соседнем атоме углерода карборана. Так, реакция 1-i-Pr2P-2-(3',5'-(2",4",6"-(i-Pr)3C6H2)2C6H3)-1,2-C2B10H10 с [(CO)2RhCl]2 в бензоле при 60°C медленно (в течение 6 сут) приводит к дикарбонильному комплексу родия [(CO)2RhCl(1-i-Pr2P-2-(3',5'-(2",4",6"-(i-Pr)3C6H2)2C6H3)-1,2-C2B10H10)], который обратимо теряет CO с образованием димерного комплекса с хлоридными мостиками [(CO)Rh(μ-Cl)(1-i-Pr2P-2-(3',5'-(2",4",6"-(i-Pr)3C6H2)2C6H3)-1,2-C2B10H10)]2 (схема 7 , рис. 8) [44].
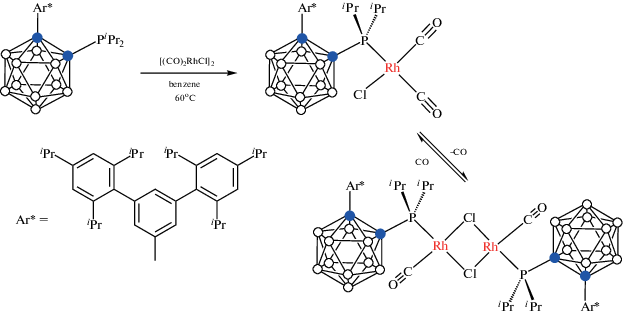
Схема 7.
Рис. 8.
Строение комплекса [(CO)Rh(μ-Cl)(1-i-Pr2P-2-(3',5'-(2",4",6"-(i-Pr)3C6H2)2C6H3)-1,2-C2B10H10)]2 (атомы водорода заместителей не показаны для ясности).
Взаимодействие 1-Ph2PCH2-1,2-C2B10H11 с RuCl3 в 2-метоксиэтаноле в присутствии формальдегида приводит к дифосфиновому комплексу [RuCl(CO)(1-Ph2PCH2-1,2-C2B10H10-κ2-P,B(3))(1-Ph2PCH2-1,2-C2B10H11-κ2-P,H)], в котором один карборанилфосфиновый лиганд координирован по κ2-P,B-типу, а второй – по κ2-P,HB-типу. Реакция этого комплекса с CO в бензоле приводит к замене BH-группы карборана в координационной сфере металла карбонилом с образованием комплекса [RuCl(CO)2(1-Ph2PCH2-1,2-C2B10H10-κ2-P,B(3))(1-Ph2PCH2-1,2-C2B10H11-κ1-P)], в котором оба фосфиновых лиганда находятся в транс-положении и который медленно изомеризуется в соответствующий цис-изомер (рис. 9, схема 8 ) [45, 46]. Взаимодействие [RuCl(CO)(1-Ph2PCH2-1,2-C2B10H10-κ2-P,B(3))(1-Ph2PCH2-1,2-C2B10H11-κ2-P,H)] с более сильными лигандами, такими как 2,2'-бипиридин или $4$-пиколин, приводит к полной потере одного из карборанилфосфиновых лигандов с образованием комплексов [RuCl(CO)(1-Ph2PCH2-1,2-C2B10H10-κ2-P,B(3))(NC5H4-4-Me)2] и [RuCl(CO)(1-Ph2PCH2-1,2-C2B10H10-κ2-P,B(3))(2,2'-Bipy)] соответственно (схема 8 ) [45].
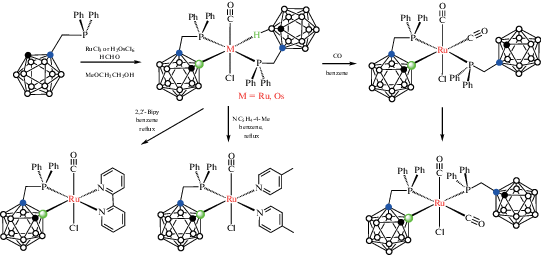
Схема 8.
Рис. 9.
Строение комплекса [RuCl(CO)2(1-Ph2PCH2-1,2-C2B10H10-κ2-P,B(3))(1-Ph2PCH2-1,2-C2B10H11-κ1-P)] (атомы водорода не показаны для ясности).
Реакция 1-Ph2PCH2-1,2-C2B10H11 с H3OsCl6 протекает аналогичным образом, приводя к [OsCl(CO)(1-Ph2PCH2-1,2-C2B10H10-κ2-P,B(3))(1-Ph2PCH2-1,2-C2B10H11-κ2-P,H)] (схема 8 ), в то время как в случае стерически затрудненного карборанилфосфина 1-Ph2PCH2-2-Me-1,2-C2B10H10 реакция дает смесь продуктов B(3)- и B(4)-активации [46].
В нидо-карборанилфосфине [7-Ph2P-7,8-C2B9H11]–, образующемся при удалении одной из соседних с атомами углерода борных вершин 1-дифенилфосфино-орто-карборана, три соседние с замещенным атомом углерода BH-группы (положения 2, 3 и 11) сильно отличаются по степени своей гидридности. При этом в отсутствие других лигандов наиболее гидридная из них может выступать в качестве донора при образовании комплексов с переходными металлами. Так, реакция [7-Ph2P-8-Ph-7,8-C2B9H10]– с циклометаллированным палладиевым комплексом [Pd((R)-C6H4CHMeNMe2-κ2-C,N)(μ-Cl)]2 в хлористом метилене приводит к смеси диастереомерных комплексов со связью Pd…H–B [Pd((R)-C6H4CHMeNMe2-κ2-C,N)((R/S)-7-Ph2P-8-Ph-7,8-C2B9H10-κ2-P,H))] (схема 9 , рис. 10), которые были разделены дробной кристаллизацией [47].
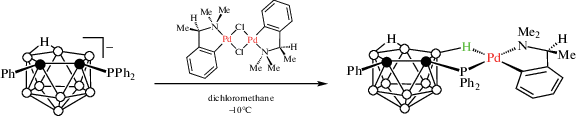
Схема 9.
Рис. 10.
Строение комплекса [Pd((R)-C6H4CHMeNMe2-κ2-C,N)((R)-7-Ph2P-8-Ph-7,8-C2B9H10-κ2-P,H)] (атомы водорода органических заместителей и лигандов не показаны для ясности).
Ряд плоскоквадратных комплексов родия [Rh(7-R2P-8-R'-7,8-C2B9H10-κ2-P,H)(PPh3)2] (R = Ph, R' = H, Me, Ph; R = Et, R' = Me, Ph; R = i-Pr, R' = = Me), в которых карборанилфосфиновый лиганд координируется металлом с помощью связей P–Rh и B(11)–H…Rh, был получен взаимодействием соответствующих нидо-карборанилфосфинов [7-R2P-8-R'-7,8-C2B9H10]– с [(Ph3P)3RhCl] в этаноле (схема 10 , рис. 11) [48]. В то же время взаимодействие нидо-карборанилфосфинов [7-Ph2P-8-R'-7,8-C2B9H10]– (R' = = H, Me, Ph) с [(COD)RhCl]2 в хлористом метилене или этаноле приводит к соответствующим пятикоординационным комплексам родия [(COD)Rh(7-Ph2P-8-R'-7,8-C2B9H10-κ3-P,H,H')], в которых карборанилфосфиновый лиганд координируется родием через PPh2-, B(11)–H- и B(2)–H-группы (схема 10 , рис. 11) [47, 49]. Реакции рацемического или энантиомерно чистого нидо-карборанилфосфина [7-Ph2P-8-Ph-7,8-C2B9H10]– с [(COD)RhCl]2 в присутствии хелатных лигандов бис(фенилфосфино) (1,1'-бис(дифенилфосфино)ферроцен, 1,4-бис(дифенилфосфино)бутан, (S)-BINAP, (R,R)- или (S,S)-DIOP) в этаноле приводят к соответствующим плоскоквадратным комплексам родия [Rh(7-Ph2P-8-Ph-7,8-C2B9H10-κ2-P,H){(Ph2P)2Z-κ2-P,P'}], где (Ph2P)2Z = = Fe(C5H4PPh2), Ph2P(CH2)4PPh2, BINAP или DIOP (схема 10 ) [47].
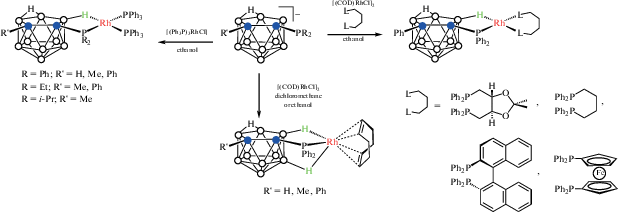
Схема 10.
Рис. 11.
Строение комплексов [Rh(7-Ph2P-7,8-C2B9H11-κ2-P,H)(PPh3)2] (а), [(COD)Rh(7-Ph2P-8-Me-7,8-C2B9H10-κ3-P,H,H')] (б) и [(COD)Rh(7-Ph2P-8-Ph-7,8-C2B9H10-κ3-P,H,H')] (в). Атомы водорода органических заместителей и лигандов не показаны для ясности.
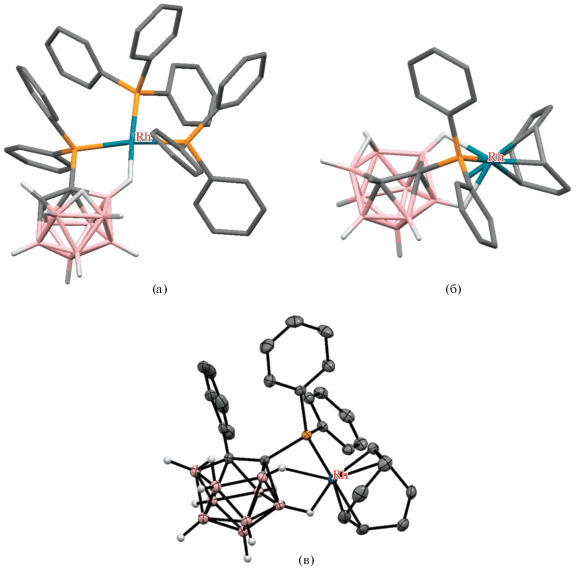
Благодаря высокой лабильности связей B–H…Rh экзо-нидо-карборанилфосфиновые комплексы родия проявляют высокую каталитическую активность в различных реакциях [50]. Так, комплексы [Rh(7-Ph2P-8-R-7,8-C2B9H10-κ2-P,H)(PPh3)2] (R = H, Me) катализируют реакцию гидрирования метациклина до доксициклина (антибиотика широкого спектра действия) с высоким выходом и очень высокой диастереоселективностью (~100%) [48]. Как [Rh(7-Ph2P-8-R-7,8-C2B9H10-κ2-P,H)(PPh3)2] (R = H, Me), так и [(COD)Rh(7-Ph2P-8-Me-7,8-C2B9H10-κ3-P,H,H')] являются активными катализаторами циклопропанирования олефинов диазоацетатами [51]. Комплексы [Rh(7-R2P-8-R'-7,8-C2B9H10-κ2-P,H)(PPh3)2] (R = = Ph, R' = H, Me; R = Et, R' = Me; R = i-Pr, R' = = Me) были изучены в качестве катализаторов реакции гидрирования 1-гексена [48], а хиральные комплексы [(COD)Rh((R)- и (S)-7-Ph2P-8-Ph-7,8-C2B9H10-κ3-P,H,H')] и [Rh((R)- и (S)-7-Ph2P-8-Ph-7,8-C2B9H10-κ2-P,H)((R,R)-DIOP-κ2-P,P')] исследовались как катализаторы энантиоселективного гидрирования ацетамидоянтарной кислоты и кетопантолактона, а также энантиоселективного гидросилилирования ацетофенона [47].
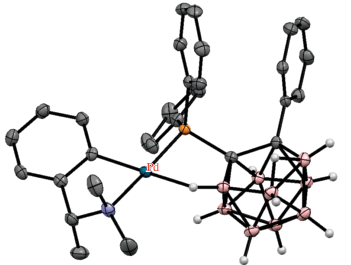
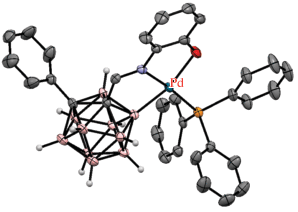
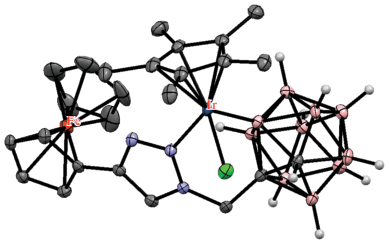
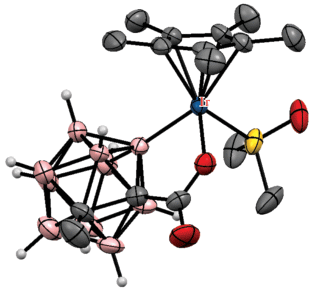
Иридиевый комплекс [(η5-C5Me5)IrH(7-Ph2P-7,8-C2B9H11-κ2-P,H)] (рис. 12) был получен непосредственно реакцией 1-дифенилфосфино-орто-карборана с [Cp*IrCl2]2 в метаноле в присутствии ацетата натрия (схема 11 ) [52].
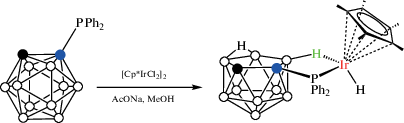
Схема 11.
Рис. 12.
Строение комплекса [(η5-C5Me5)IrH(7-Ph2P-7,8-C2B9H11-κ2-P,H)] (атомы водорода органических заместителей и лигандов не показаны для ясности).
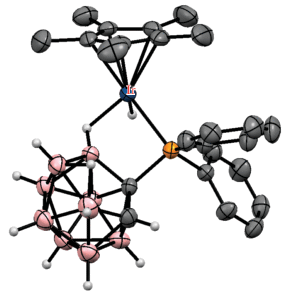
Рис. 13.
Строение различных изомеров комплекса [Ru(7-Ph2P-8-Me-7,8-C2B9H10-κ3-P,H,H')2] (атомы водорода органических заместителей и лигандов не показаны для ясности).
Взаимодействие [7-Ph2P-8-Me-7,8-C2B9H10]– с RuCl3 ‧ nH2O или [RuCl2(DMSO)4] приводит к образованию бис(фосфинового) комплекса [Ru(7-Ph2P-8-Me-7,8-C2B9H10-κ3-P,H,H')2], в котором оба нидо-карборанилфосфиновых лиганда координируются атомом рутения через PPh2-, B(11)–H- иB(2)–H-группы (схема 12 ). Вследствие наличия в исходном нидо-карборанилфосфине двух энантиомеров такая координация может приводить к образованию шести различных изомеров, два из которых были охарактеризованы с помощью рентгеноструктурного анализа (рис. 13 ) [53].
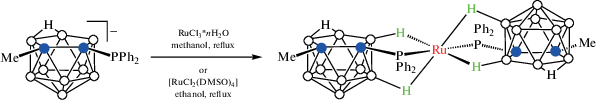
Схема 12.
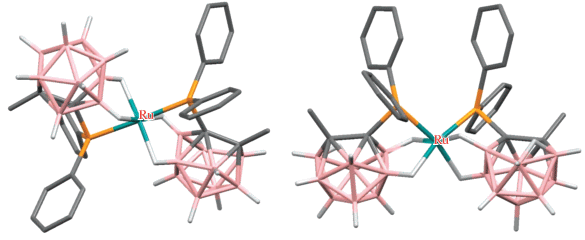
Взаимодействие нидо-карборанилфосфинов [7-R2P-8-R'-7,8-C2B9H10]– (R = Et, R' = Et; R = Ph, R' = H, Me, Ph) с [RuCl2(PPh3)3] в кипящем этаноле приводит к соответствующим октаэдрическим комплексам рутения [RuCl(7-R2P-8-R'-7,8-C2B9H10-κ3-P,H,H')(PPh3)2], в которых карборанилфосфиновый лиганд координируется к атому рутения через PRh2-, B(11)–H- и B(2)–H-группы (схема 13 , рис. 14). Трифенилфосфиновый лиганд в транс-положении к PRh2-группе может быть легко замещен молекулой этанола, тетрагидротиофена или оксида углерода с образованием соответствующих комплексов [RuCl(7-R2P-8-R'-7,8-C2B9H10-κ3-P,H,H')(PPh3)(L)] (L = CO, THT, EtOH) (схема 13 , рис. 14). Аналогичным образом реакции нидо-карборанилфосфинов [7-Ph2P-8-R-7,8-C2B9H10]– (R = H, Me, Ph) с [RuH(AcO)Cl2(PPh3)3] в кипящем этаноле приводят к соответствующим гидридным комплексам [RuH(7-Ph2P-8-R'-7,8-C2B9H10-κ3-P,H,H')(PPh3)2] (схема 13 ) [54]. Обнаружено, что гидридные комплексы [RuH(7-Ph2P-8-R-7,8-C2B9H10-κ3-P,H,H')(PPh3)2] (R = H, Me) являются эффективными катализаторами циклопропанирования олефинов этилдиазоацетатом [55].
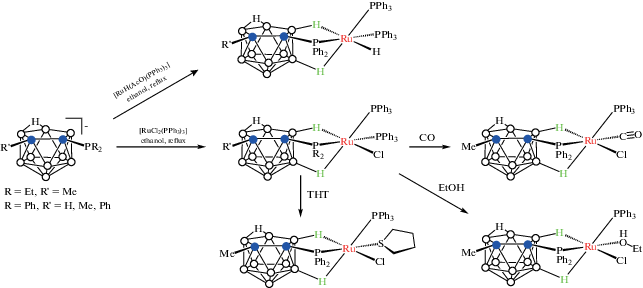
Схема 13.
Рис. 14.
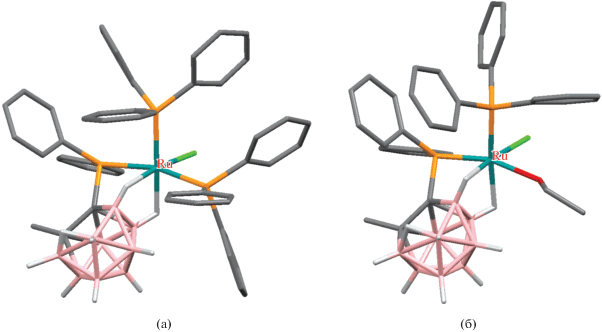
Строение комплексов [RuCl(7-Ph2P-8-Me-7,8-C2B9H10-κ3-P,H,H')(PPh3)2] (а) и [RuCl(7-Ph2P-8-Me-7,8-C2B9H10-κ3-P,H,H')(PPh3)(EtOH)] (б). Атомы водорода органических заместителей и лигандов не показаны для ясности.
Интересно, что взаимодействие бис(дифенилфосфино) нидо-карборана [7,8-(Ph2P)2-7,8-C2B9H10]– с [RuCl2(PPh3)3] или [RuH(AcO)Cl2(PPh3)3] в кипящем метаноле вместо ожидаемых κ2-P,P'-комплексов приводит к аналогичным κ3-P,H,H'-комплексам, тогда как реакция с [RuCl2(PPh2Me)4] дает κ3-P,P',H-комплекс [Ru(7,8-(Ph2P)2-7,8-C2B9H10-κ3-P,P',H)2] (схема 14 ) [56].
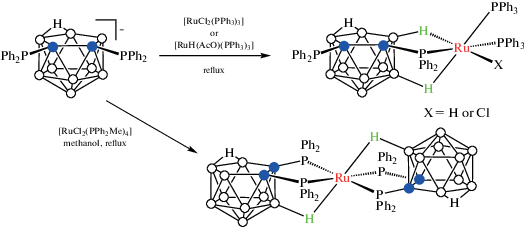
Схема 14.
Полученные рутениевые комплексы являются эффективными катализаторами присоединения четыреххлористого углерода к олефинам и радикальной полимеризации метилметакрилата и стирола [57].
Особый интерес представляют C,C'-дифосфины на основе мета-карборана, отличающиеся от C,C'-дифосфинов на основе орто-карборана, которые обычно образуют P,P'-хелатные комплексы. Для дифосфинов на основе мета-карборана положения B(2) и B(3) доступны для активации с участием обоих фосфиновых групп, что позволяет получать карборановые аналоги C-пинцетных комплексов переходных металлов. Так, реакция 1,7-бис(фосфинит)карборана 1,7-(i-Pr2PO)2-1,7-C2B10H10 с NiCl2 в смеси тетрагидрофурана и толуола при 90°C приводит к селективной B(2)–H-активации карборанового остова с образованием (POBOP)NiCl комплекса [NiCl(1,7-(i-Pr2PO)2-1,7-C2B10H9-κ3-P,B,P')]. Попытка окисления этого комплекса 1 экв. иода в тетрагидрофуране наряду с замещением хлорида иодидом приводит к активации связи B(3)–H с образованием комплекса [NiI(1,7-(i-Pr2PO)2-3-I-1,7-C2B10H8-κ3-P,B,P')] (схема 15 , рис. 15). Реакция с избытком иода приводит к окислению фосфинитных заместителей с деструкцией комплекса и образованием [1,7-(i-Pr2P(I)O)2-2,3-I2-1,7-C2B10H8](I3)2 [58].
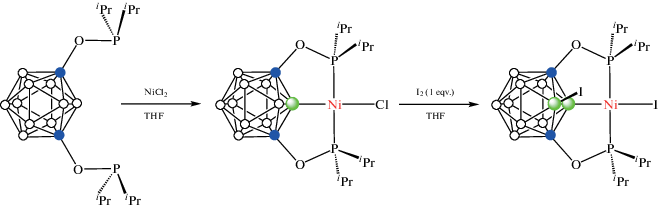
Схема 15.
Рис. 15.
Строение комплексов [NiCl(1,7-(i-Pr2PO)2-1,7-C2B10H9-κ3-P,B,P')] (а) и [NiI(1,7-(i-Pr2PO)2-3-I-1,7-C2B10H8-κ3-P,B,P')] (б). Атомы водорода не показаны для ясности.
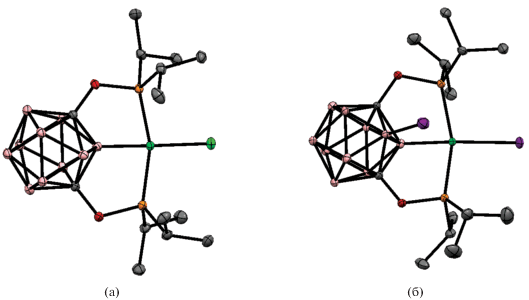
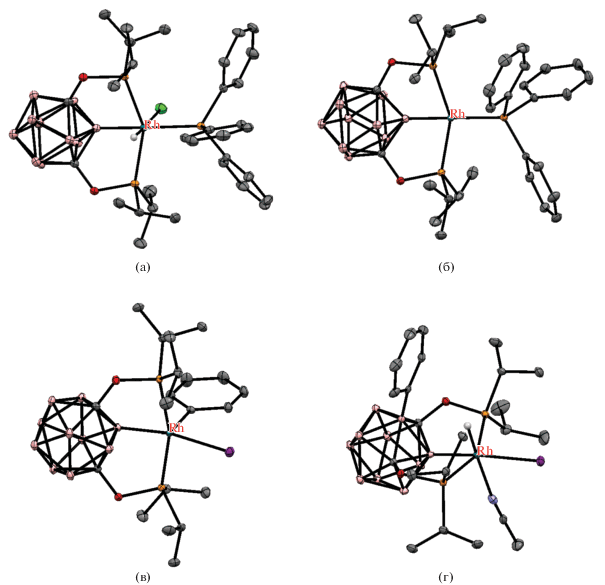
Реакция 1,7-(i-Pr2PO)2-1,7-C2B10H10 с [(Ph3P)3RhCl] в тетрагидрофуране при комнатной температуре приводит к получению POBOP пинцетного комплекса родия(III) [RhHCl(1,7-(i-Pr2PO)2-1,7-C2B10H9-κ3-P,B,P')(PPh3)], который при обработке Et3N претерпевает восстановительное элиминирование HCl с образованием 16-электронного комплекса родия(I) [RhHCl(1,7-(i-Pr2PO)2-1,7-C2B10H9-κ3-P,B,P')(PPh3)]. Последний при обработке иодбензолом в тетрагидрофуране вступает в реакцию окислительного присоединения с образованием [Rh(Ph)I(1,7-(i-Pr2PO)2-1,7-C2B10H9-κ3-P,B,P')], который, в свою очередь, при нагревании с ацетонитрилом в бензоле подвергается B(3)–H-активации с миграцией фенильной группы и образованием гидридного комплекса [RhHI(1,7-(i-Pr2PO)2-3-Ph-1,7-C2B10H8-κ3-P,B,P')(MeCN)] (схема 16 , рис. 16) [59].
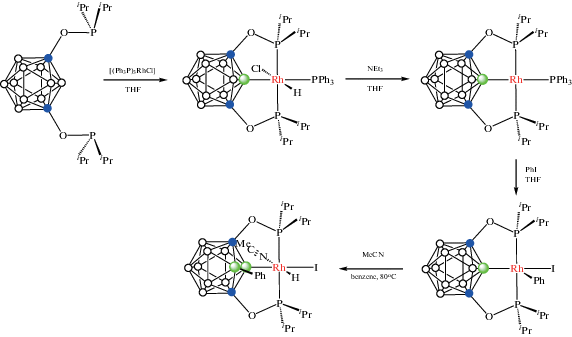
Схема 16.
Рис. 16.
Строение комплексов [RhHCl(1,7-(i-Pr2PO)2-1,7-C2B10H9-κ3-P,B,P')(PPh3)] (а), [Rh(1,7-(i-Pr2PO)2-1,7-C2B10H9-κ3-P,B,P')(PPh3)] (б), [Rh(Ph)I(1,7-(i-Pr2PO)2-1,7-C2B10H9-κ3-P,B,P')] (в) и [RhHI(1,7-(i-Pr2PO)2-3-Ph-1,7-C2B10H8-κ3-P,B,P')(MeCN)] (г). Атомы водорода не показаны для ясности.
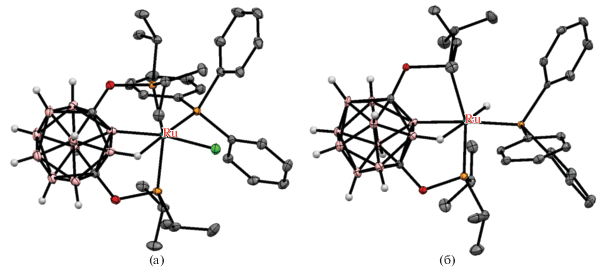
Реакция 1,7-(i-Pr2PO)2-1,7-C2B10H10 c [Ru(CO)3Cl2]2 в кипящем бензоле приводит к образованию POBOP комплекса рутения(II) [RuCl(1,7-(i-Pr2PO)2-1,7-C2B10H9-κ3-P,B,P')(CO)2], обработка которого Et3N дает (BB)-карбориновый комплекс рутения(0) [Ru(1,7-(i-Pr2PO)2-1,7-C2B10H8-κ4-P,B,B',P')(CO)2] как продукт двойной B–H-активации (рис. 17). Полученный металлоцикл (BB)>Ru может рассматриваться как неорганический аналог циклопропана с двумя сильно напряженными (2c–2e) σ-связями B–Ru. Значительное искажение экзополиэдрических связей карборана приводит к повышенной реакционной способности этих изогнутых связей B–Ru, которые могут выступать в качестве нуклеофильных центров. Поэтому реакция образования (BB)-карборинового комплекса обратима и добавление HCl приводит к разрыву одной из связей Ru–B с образованием хлоридного комплекса рутения(II) (схема 17 ). Аналогичным образом реакция (BB)-карборинового комплекса рутения(0) с терминальным ацетиленом приводит к формальному окислительному присоединению с образованием B-карборанилацетилидного комплекса рутения(II) [Ru(C≡CCO2Et)(1,7-(i-Pr2PO)2-1,7-C2B10H9-κ3-P,B,P')(CO)2], тогда как реакция с 3-гексином при УФ-облучении приводит к циклоприсоединению последнего с образованием комплекса рутения(II) с B–C(Et)=C(Et)–Ru мостиком [Ru(1,7-(i-Pr2PO)2-3-C(Et)=C(Et)-1,7-C2B10H9-κ4-P,B,C,P')(CO)2] (схема 17 , рис. 18). При взаимодействии (BB)-карборинового комплекса рутения(0) с иодом образуется комплекс рутения(II) [RuI(1,7-(i-Pr2PO)2-3-I-1,7-C2B10H8-κ3-P,B,P')(CO)], в котором связанный с соседним атомом бора атом иода дополняет координационную сферу рутения до октаэдра (схема 17 , рис. 19). Реакция (BB)-карборинового комплекса с избытком H3B · SMe2 в смеси тетрагидрофурана и бензола при 80°C приводит к комплексу [RuI(1,7-(i-Pr2PO)2-3-BH3-1,7-C2B10H8-κ4-P,B,H,P')(CO)2], содержащему экзополиэдрическую BH3-группу, которая участвует в B(3)–BH2–H···Ru-взаимодействии с атомом металла (схема 17 , рис. 19) [60].
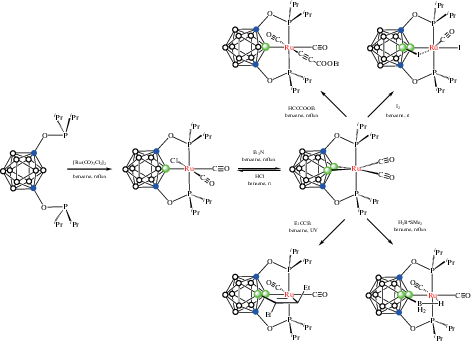
Схема 17.
Рис. 17.
Строение комплексов [RuCl(1,7-(i-Pr2PO)2-1,7-C2B10H9-κ3-P,B,P')(CO)2] (а) и [Ru(1,7-(i-Pr2PO)2-1,7-C2B10H8-κ4-P,B,B',P')(CO)2] (б). Атомы водорода не показаны для ясности.
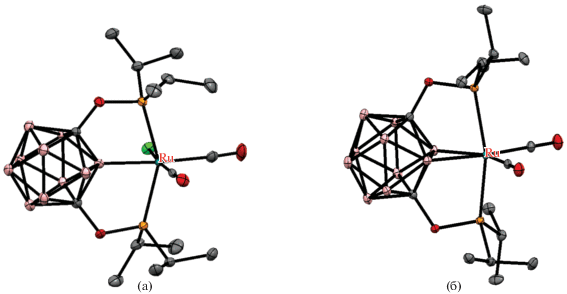
Рис. 18.
Строение комплексов [Ru(C≡CCO2Et)(1,7-(i-Pr2PO)2-1,7-C2B10H9-κ3-P,B,P')(CO)2] (а) и [Ru(1,7-(i-Pr2PO)2-3-C(Et)=C(Et)-1,7-C2B10H9-κ4-P,B,C,P')(CO)2] (б). Атомы водорода не показаны для ясности.
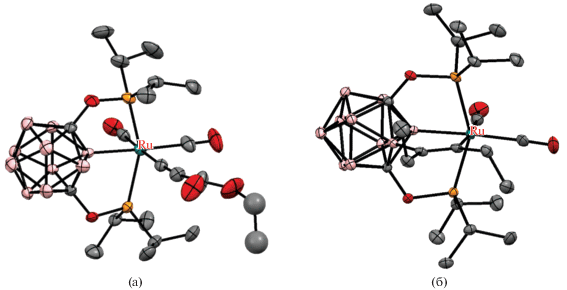
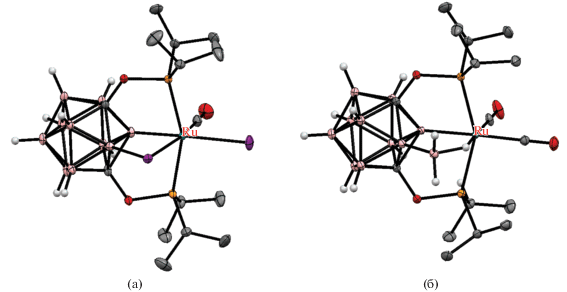
Рис. 19.
Строение комплексов [RuI(1,7-(i-Pr2PO)2-3-I-1,7-C2B10H8-κ3-P,B,P')(CO)] (а) и [RuI(1,7-(i-Pr2PO)2-3-BH3-1,7-C2B10H8-κ4-P,B,H,P')(CO)2] (б). Атомы водорода органических заместителей не показаны для ясности.
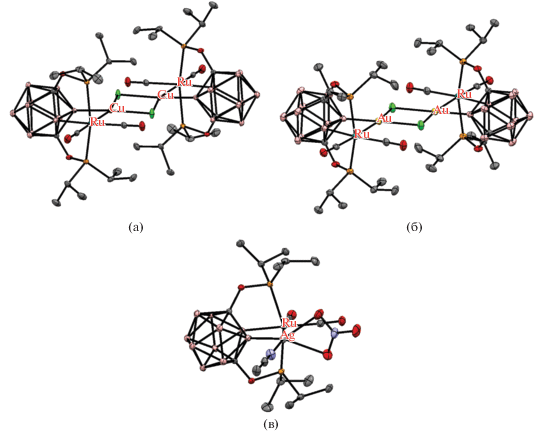
Карбориновый комплекс рутения также может взаимодействовать с неорганическими электрофилами, такими как катионы металлов подгруппы меди. Так, его реакция с CuCl в кипящей смеси тетрагидрофурана и бензола приводит к димерному комплексу [(CO)2Ru-Cu(μ-Cl)(1,7-(i-Pr2PO)2-1,7-C2B10H8-κ4-P,B,B',P')]2 с четырехчленным металлоциклом B–Cu–Ru–B (рис. 20). Аналогичный комплекс с B–Au–Ru–B-металлоциклом [(CO)2Ru-Au(μ-Cl)(1,7-(i-Pr2PO)2-1,7-C2B10H8-κ4-P,B,B',P')]2 (рис. 20) был получен реакцией с [(Me2S)AuCl] в хлористом метилене при комнатной температуре. В обоих комплексах катионы Cu+ и Au+ находятся в необычном плоскоквадратном окружении. Реакция карборинового комплекса с AgNO3 в смеси хлористого метилена и бензола после кристаллизации из ацетонитрила дает мономерный биметаллический комплекс [(CO)2Ru-Ag(MeCN)(η2-NO3)(1,7-(i-Pr2PO)2-1,7-C2B10H8-κ4-P,B,B',P')] (рис. 20) [61].
Рис. 20.
Строение комплексов [(CO)2Ru-Cu(μ-Cl)(1,7-(i-Pr2PO)2-1,7-C2B10H8-κ4-P,B,B',P')]2 (а), [(CO)2Ru-Au(μ-Cl)(1,7-(i-Pr2PO)2-1,7-C2B10H8-κ4-P,B,B',P')]2 (б) и [(CO)2Ru-Ag(MeCN)(η2-NO3)(1,7-(i-Pr2PO)2-1,7-C2B10H8-κ4-P,B,B',P')] (в). Атомы водорода не показаны для ясности.
Взаимодействие 1,7-(i-Pr2PO)2-1,7-C2B10H10 с [RuCl2(PPh3)3] в кипящем тетрагидрофуране приводит к образованию POBOP пинцетного комплекса рутения(II) [RuCl(1,7-(i-Pr2PO)2-1,7-C2B10H9-κ4-P,B,P,H')(PPh3)], который при обработке NaH в тетрагидрофуране дает соответствующий гидридный комплекс [RuH(1,7-(i-Pr2PO)2-1,7-C2B10H9-κ4-P,B,P',H)(PPh3)], в котором имеются связи B–Ru, B–H…Ru и Ru–H (схема 18 , рис. 21). В этом комплексе наблюдается быстрый обмен между атомами водорода карборана и концевого гидрида вследствие миграции атома металла между положениями B(2) и B(3) карборанового остова с барьером активации 12.2 ккал/моль. Образующая контакт B–H…Ru группа B(3)H играет роль полулабильного лиганда, защищающего вакантное координационное место в комплексе [RuH(1,7-(i-Pr2PO)2-1,7-C2B10H9-κ4-P,B,P',H)(PPh3)], который является хорошим катализатором дегидрирования циклооктана [62].
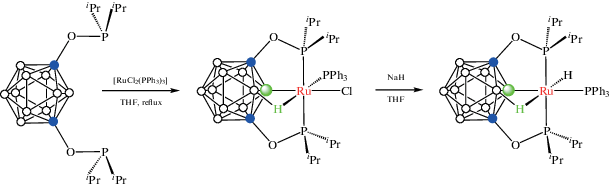
Схема 18.
Рис. 21.
Строение комплексов [RuCl(1,7-(i-Pr2PO)2-1,7-C2B10H9-κ4-P,B,P,H')(PPh3)] (а) и [RuH(1,7-(i-Pr2PO)2-1,7-C2B10H9-κ4-P,B,P,H')(PPh3)] (б). Атомы водорода органических заместителей и лигандов не показаны для ясности.
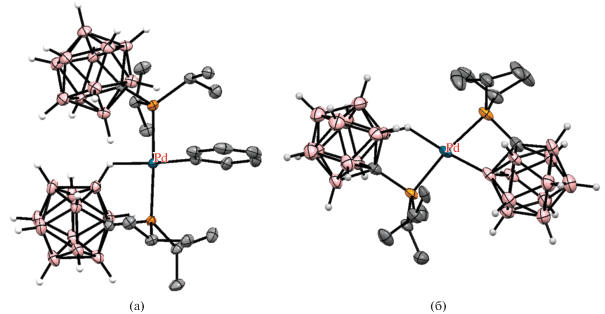
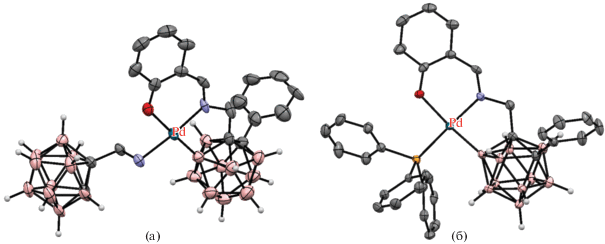
Карборанилфосфины на основе карба-клозо-додекаборатного аниона [CB11H12]– ведут себя во многом аналогично карборанилфосфинам на основе орто-карборана. Так, реакция [Li(THF)3][1-i-Pr2P-1-CB11H11] с [(COD)Pd(CH2SiMe3)2] в бензоле приводит к 14-электронному комплексу палладия(0) [Li(THF)4]2[Pd(1-i-Pr2P-1-CB11H11)2], который при комнатной температуре реагирует с хлорбензолом с образованием комплекса палладия(II) [Li(THF)(Et2O)3][Pd(Ph)(1-i-Pr2P-1-CB11H11-κ1-P)(1-i-Pr2P-1-CB11H11-κ2-P,H)], в котором BH-группа играет роль одного из лигандов в плоскоквадратном окружении палладия (рис. 22). Реакция с хлорбензолом при 80°C приводит к элиминированию бензола с образованием циклометаллированного комплекса [Li(THF)4][Pd(1-i-Pr2P-1-CB11H11-κ2-P,H)(1-i-Pr2P-1-CB11H10-κ2-P,B)] (схема 19 , рис. 22) [63, 64].
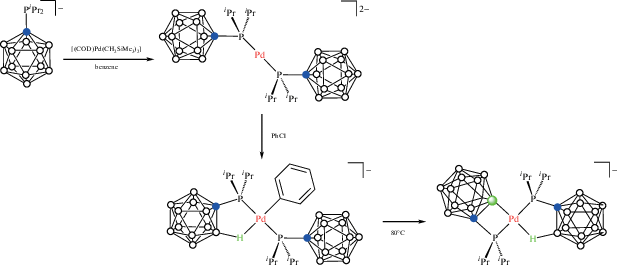
Схема 19.
Рис. 22.
Строение комплексов [Pd(Ph)(1-i-Pr2P-1-CB11H11-κ1-P)(1-i-Pr2P-1-CB11H11-κ2-P,H)]– (а) и [Pd(1-i-Pr2P-1-CB11H11-κ2-P,H)(1-i-Pr2P-1-CB11H10-κ2-P,B)]– (б). Атомы водорода органических заместителей и лигандов не показаны для ясности.
Реакция [Li(THF)3][1-i-Pr2P-1-CB11H11] с [Rh(CO)2Cl]2 и AgBF4 в хлористом метилене приводит к нестабильному комплексу Li[Rh(1-i-Pr2P-1-CB11H11)2(CO)2], который легко отщепляет один лиганд CO с образованием комплекса Li[Rh(1-i-Pr2P-1-CB11H11-κ1-P)(1-i-Pr2P-1-CB11H11-κ2-P,H)(CO)], в котором одна из BH-групп карборана играет роль лиганда в плоскоквадратном окружении родия (схема 20 , рис. 23).
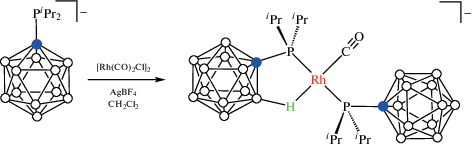
Схема 20.
Рис. 23.
Строение комплекса [Rh(1-i-Pr2P-1-CB11H11-κ1-P)(1-i-Pr2P-1-CB11H11-κ2-P,H)(CO)]– (атомы водорода органических заместителей не показаны для ясности).
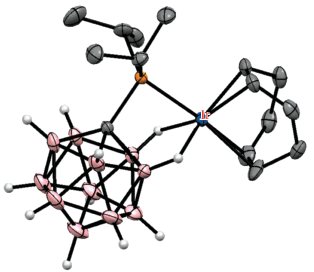
Реакция [Li(THF)3][1-i-Pr2P-1-CB11H11] с [Ir(COD)Cl]2 в бензоле приводит к образованию комплекса [(COD)Ir(1-i-Pr2P-1-CB11H11-κ3-P,H,H)], в котором атом иридия дополнительно координируется двумя BH-группами карборанового остова (рис. 24) [65].
Реакции B–H-активации с участием азотсодержащих направляющих лигандов
Благодаря разнообразию азотсодержащих заместителей, которые могут выступать в качестве направляющих лигандов, реакции B–H-активации с их участием являются одними из наиболее изученных. Первыми из них были описаны реакции 1-Me2NCH2-2-Ph-1,2-C2B10H10 с метилпентакарбонилмарганцем [(CO)5MnMe] и метилпентакарбонилрением [(CO)5ReMe] при нагревании в углеводородных растворителях, приводящие к продуктам циклометаллирования со связью бор–металл [M(1-Me2NCH2-2-Ph-1,2-C2B10H9-κ2-N,B)(CO)4] (M = Mn, Re) [66, 67]. Положение B–H-активации при этом не установлено.
Реакции 1-диметиламинометил-7-фенил-мета-карборана 1-Me2NCH2-7-Ph-1,7-C2B10H10 с [(CO)5MMe] (M = Mn, Re) протекают аналогично, приводя к циклометаллированным комплексам [(CO)4M(1-Me2NCH2-7-Ph-1,7-C2B10H9-κ2-N,B)] (M = Mn, Re) [66, 67]. Согласно данным рентгеноструктурного анализа [(CO)4Re(1-Me2NCH2-7-Ph-1,7-C2B10H9-κ2-N,B(4))] (рис. 25), B–H-активация протекает по положению 4, что объясняется затрудненностью замещения по положению 2, вызванной фенильным заместителем при соседнем атоме углерода [68].
Рис. 25.
Строение комплекса [(CO)4Re(1-Me2NCH2-7-Ph-1,7-C2B10H9-κ2-N,B(4))] (атомы водорода не показаны для ясности).
Взаимодействие 1-R2NCH2-2-R'-1,2-C2B10H10 (R = Me, R' = Ph; R = Et, R' = H) с [(PhCN)2PdCl2] в бензоле при комнатной температуре приводит к образованию соответствующих циклопалладированных димерных комплексов [PdCl(1-R2NCH2-2-R'-1,2-C2B10H9-κ2-N,B)]2, обработка которых 4-метилпиридином протекает с разрывом дихлоридного мостика и образованием комплексов [PdCl(1-R2NCH2-2-R'-1,2-C2B10H9-κ2-N,B)(NC5H4-4-Me)]. Связь B–Pd в этих комплексах легко разрывается под действием кислот. Реакция как [PdCl(1-Et2NCH2-1,2-C2B10H10-κ2-N,B)]2, так и [PdCl(1-Et2NCH2-1,2-C2B10H10-κ2-N,B)(NC5H4-4-Me)] с дифенилацетиленом приводит к внедрению двух молекул в связь бор–металл с образованием пятичленного металлоцикла, в котором атом палладия координирован двойной связью бутадиенового фрагмента [PdCl(1-Et2NCH2-X-C(Ph)=C(Ph)-C(Ph)=C(Ph)-1,2-C2B10H10-η2-(C(C(1')=C(2'))-κ2-N,C(4'))] [69].
Аналогичным образом реакция 1-Me2NCH2-7-Ph-1,7-C2B10H10 с [(PhCN)2PdCl2] дает соответствующий циклопалладированный димер [PdCl(1-Me2NCH2-7-Ph-1,7-C2B10H9-κ2-N,B)]2, взаимодействие которого с 4-метилпиридином приводит к [PdCl(1-Me2NCH2-7-Ph-1,7-C2B10H9-κ2-N,B)(NC5H4-4-Me)] [69].
Реакция 1-аминометил-2-фенил-орто-карборана 1-NH2CH2-2-Ph-1,2-C2B10H10 с арилиодидами в присутствии 10 мол. % Pd(OAc)2, 20 мол. % глиоксалевой кислоты, CF3COOAg и уксусной кислоты в гексафторизопропаноле при 60°C приводит к образованию B(4,5)-диарилпроизводных 1-NH2CH2-2-Ph-4,5-Ar2-1,2-C2B10H8 (схема 21 ). Наличие электроноакцепторных или электронодонорных заместителей не оказывает большого влияния на выход арилпроизводных, который колеблется от 50 до 87%. Вместо фенильной группы второй атом углерода в карборане также может нести замещенные арильные или алкильные группы. В качестве направляющих также могут использоваться 1-NH2CHMe- и 1-NH2CMe2-группы, а вместо глиоксалевой кислоты – салицилальдегид, 2-кетопропионовая или фенилглиоксалиевая кислота. Изучение механизма реакции показало, что взаимодействие 1-аминометил-2-фенил-орто-карборана с салицилальдегидом при комнатной температуре в присутствии Pd(OAc)2 протекает через образование основания Шиффа и приводит к образованию B(4)-циклометаллированного хелатного комплекса [Pd(1-NH2CH2-2-Ph-1,2-C2B10H10-κ-N)(1-(O-2'-C6H4CH=NCH2)-2-Ph-1,2-C2B10H9-κ3-O,N,B(4))] (рис. 26), реакция которого с иодбензолом в присутствии CF3COOAg в гексафторизопропаноле при 80°C приводит к смеси 1-(HO-2'-C6H4CH=NCH2)-2,4,5-Ph3-1,2-C2B10H8 и 1-CF3CONHCH2-2,4,5-Ph3-1,2-C2B10H8. Кроме того, было установлено, что этот комплекс, а также его аналог [Pd(PPh3)(1-(O-2'-C6H4CH=NCH2)-2-Ph-1,2-C2B10H9-κ3-O,N,B(4))] (рис. 26), образующийся при замещении карбораниламина трифенилфосфином, способны выступать в качестве катализаторов арилирования [70].
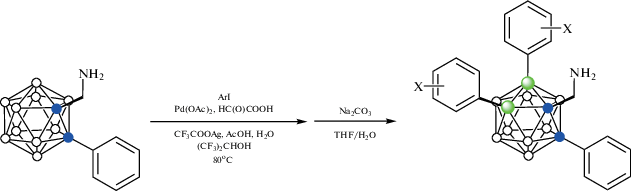
Схема 21.
Рис. 26.
Строение комплексов [Pd(1-NH2CH2-2-Ph-1,2-C2B10H10-κ-N)(1-(O-2'-C6H4CH=NCH2)-2-Ph-1,2-C2B10H9-κ3-O,N,B(4))] (а) и [Pd(PPh3)(1-(O-2'-C6H4CH=NCH2)-2-Ph-1,2-C2B10H9-κ3-O,N,B(4))] (б). Атомы водорода органических заместителей и лигандов не показаны для ясности.
Реакция 1-арилимино-орто-карборанов 1-ArN=CH-1,2-C2B10H11 (Ar = C6H4-4-F, C6H4-4-Cl, C6H2-2,4,6-Me3) с различными N-ацилглутар- и сукцинимидами RC(O)N(CO)2(CH2)n (n = 2, 3) в присутствии 5 мол. % [(COD)RhCl2]2 в толуоле при 150°С приводит к смеси соответствующих B(3)-моно- и B(3),B(6)-дизамещенных производных 1-ArN=CH-3-R-1,2-C2B10H10 и 1-ArN=CH-3,6-R2-1,2-C2B10H9 (схема 22 ). Реакция может быть использована для введения в карборановый остов арильных заместителей, содержащих различные функциональные группы, а также гетероарильных, винильных и алкильных заместителей. Следует отметить, что направляющая арилиминогруппа может быть удалена в две стадии, включая кислотный гидролиз до альдегида с последующим отрывом формильной группы при обработке перманганатом калия, что делает привлекательным ее использование в синтезе производных орто-карборана [71].
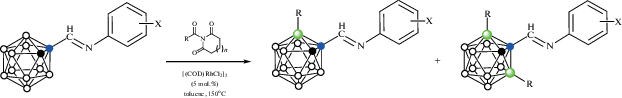
Схема 22.
Направляющая арилиминогруппа может также генерироваться in situ при взаимодействии карборанилальдегидов с аминами. Так, реакция 1-формил-2-фенил-орто-карборана 1-H(O)C-2-Ph-1,2-C2B10H10 с орто-аминофенолом и Pd(OAc)2 в толуоле в присутствии трифенилфосфина дает B(4)-циклометаллированный комплекс [(Ph3P)Pd(1-(2'-OC6H4N=CH)-2-Ph-1,2-C2B10H9-κ3-O,N,B(4))] (рис. 27). Реакция последнего с метил-пара-иодобензоатом 4-IC6H4COOMe, CF3COOAg и трифторуксусной кислотой в гексафторизопропаноле с последующим кислотным гидролизом дает смесь соответствующих B(4)-моно- и B(4,5)-диарилзамещенных карборанов 1-H(O)C-2-Ph-4-(4'-MeO(O)CC6H4)-1,2-C2B10H9 и 1-H(O)C-2-Ph-4,5-(4'-MeO(O)CC6H4)2-1,2-C2B10H8. Орто-аминофенол в этой реакции с успехом может быть заменен глицином, а избыток арилиодида приводит к продуктам B(4,5)-диарилирования (схема 23 ). Реакция хорошо протекает с арилиодидами, содержащими как донорные, так и акцепторные заместители, при этом выход продуктов колеблется от 55 до 86%. Фенильная группа при втором атоме углерода карборана может быть заменена как другими арильными (C6H4-p-Me, C6H4-p-OMe), так и бензильными (CH2Ph, CHPh2) или алкильными (Me, i-Pr) группам, однако в отсутствие заместителя реакция приводит к образованию неразделимой смеси продуктов [72].
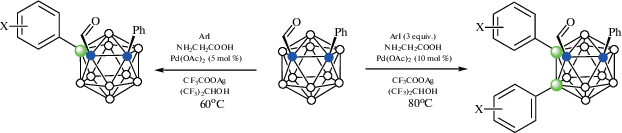
Схема 23.
Рис. 27.
Строение комплекса [(Ph3P)Pd(1-(2'-OC6H4N=CH)-2-Ph-1,2-C2B10H9-κ3-O,N,B(4))] (атомы водорода органических заместителей и лигандов не показаны для ясности).
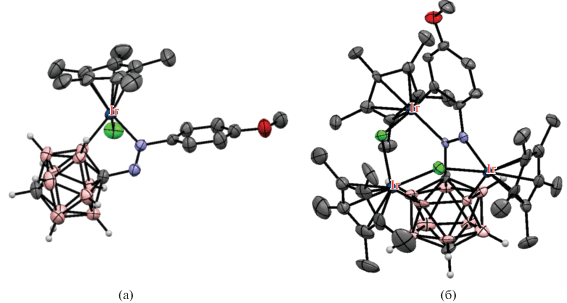
Роль направляющего лиганда может также играть арилазогруппа. Так, взаимодействие фенилазопроизводного 1-PhN=N-2-i-Pr-1,2-C2B10H10 с [(CO)5MMe] (M = Mn, Re) в углеводородных растворителях приводит к образованию соответствующих продуктов B(4)-циклометаллирования [(CO)4M(1-PhN=N-2-i-Pr-1,2-C2B10H9-κ2-N(2),B(4))] (рис. 28) [66, 67, 73].
Рис. 28.
Строение комплекса [(CO)4Re(1-PhN=N-1,2-C2B10H10-κ2-N(2),B(4))] (атомы водорода не показаны для ясности).
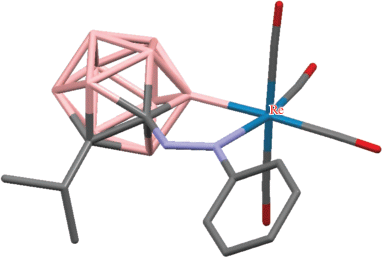
Реакция арилазопроизводных 1-(4'-RC6H4N=N)-1,2-C2B10H11 с [Cp*IrCl2]2 в хлористом метилене в присутствии NaOAc приводит к образованию соответствующих B(3)-циклометаллированных комплексов [Cp*IrCl(1-(4'-RC6H4N=N)-1,2-C2B10H10-κ2-N(2),B(3))] (R = = F, MeO) с выходом 90–94% (схема 24 , рис. 29). Аналогичная реакция с [Cp*RhCl2]2 требует использования в качестве основания n-BuLi, приводя с низким выходом к [Cp*RhCl(1-(4'-FC6H4N=N)-1,2-C2B10H10-κ2-N(2),B(3))] (рис. 29) [74].
Рис. 29.
Строение комплексов [Cp*IrCl(1-(4-MeOC6H4N=N)-1,2-C2B10H10-κ2-N(2),B(3))] (а), [Cp*IrCl(1-(4-FC6H4N=N)-1,2-C2B10H10-κ2-N(2),B(3))] (б) и [Cp*RhCl(1-(4-FC6H4N=N)-1,2-C2B10H10-κ2-N(2),B(3))] (в). Атомы водорода органических заместителей и лигандов не показаны для ясности.
При обработке PhXLi (X = S, Se) хлоридный лиганд в комплексе [Cp*IrCl(1-(MeO-RC6H4N=N)-1,2-C2B10H10-κ2-N(2),B(3))] может быть легко заменен фенилтиолатом или фенилселенолатом с образованием соответствующих комплексов [Cp*Ir(XPh)(1-(MeO-RC6H4N=N)-1,2-C2B10H10-κ2-N(2),B(3))] с почти количественным выходом. Реакция [Cp*IrCl(1-(MeO-RC6H4N=N)-1,2-C2B10H10-κ2-N(2),B(3))] с AgOTf с последующим добавлением орто-карборанилкарбоксилата приводит к [Cp*Ir(1'-OOC-1',2'-C2B10H11-κ1-O)(1-(MeO-RC6H4N=N)-1,2-C2B10H10-κ2-N(2),B(3))] (схема 24 ) [74].
Реакция 1-(4'-MeOC6H4N=N)-1,2-C2B10H11 с 1 экв. [Cp*IrCl2]2 и 2 экв. AgOTf в присутствии NaOAc в хлористом метилене приводит к образованию продукта одновременно C–H- и B–H-активации – синему комплексу [(Cp*IrCl)2(1-(4'-MeOC6H3N=N)-1,2-C2B10H10-κ4-N(2),B(3);N(1),C(2'))] (рис. 30) с выходом 80%, а аналогичная реакция с 2 экв. [Cp*IrCl2]2 и 4 экв. AgOTf дает с выходом 85% продукт C–H- и двойной B–H-активации – коричневый катионный комплекс [(Cp*Ir)3(μ-Cl)2(1-(4'-MeOC6H3N=N)-1,2-C2B10H9-κ5-N(2),B(3); N(1),C(2');B(4))]OTf, в котором третий фрагмент Cp*Ir координирован атомом бора в положении 4 и двумя мостиковыми хлоридными лигандами (схема 24 ) [75].
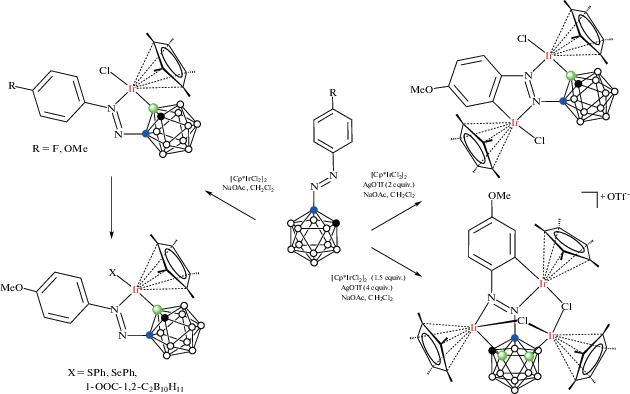
Схема 24.
Рис. 30.
Строение комплекса [(Cp*IrCl)2(1-(4'-MeOC6H3N=N)-1,2-C2B10H10-κ4-N(2),B(3);N(1),C(2'))] (атомы водорода органических заместителей и лигандов не показаны для ясности).
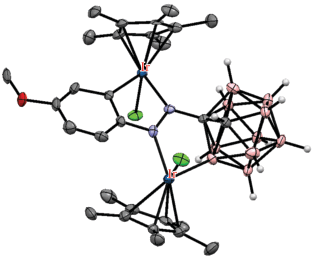
Реакция 1,2-бис(арилазо)производного орто-карборана 1,2-(4'-MeOC6H4N=N)2-1,2-C2B10H10 с 0.5 экв. [Cp*IrCl2]2 и Et3N в хлористом метилене приводит с выходом 75% к комплексу [Cp*IrCl(1-(4'-MeOC6H4N=N)2-1,2-C2B10H9-κ2-N(2),B(4))], а реакция с 1 экв. [Cp*IrCl2]2 – к биядерному комплексу [(Cp*IrCl)2(1-(4'-MeOC6H4N=N)2-1,2-C2B10H8-κ4-N(2),B(4);N(2'),B(7))] с выходом 65% [76].
Фенилазопроизводное мета-карборана 1-PhN=N-7-Me-1,7-C2B10H10 реагирует с [(CO)5MnMe] и [(CO)5ReMe] аналогично фенилазопроизводному орто-карборана с образованием соответствующих циклометаллированных комплексов [(CO)4M(1-PhN=N-7-Me-1,7-C2B10H9-κ2-N,B)] (M = Mn, Re) [66, 67].
Реакция арилазопроизводного пара-карборана 1-(4'-MeOC6H4N=N)-1,12-C2B10H11 с [Cp*IrCl2]2 в хлористом метилене в присутствии NaOAc приводит к образованию соответствующего B(2)-циклометаллированного комплекса [Cp*IrCl(1-(4'-MeOC6H4N=N)-1,12-C2B10H10-κ2-N(2),B(2))] (рис. 31) с выходом 92%, а реакция с избытком [Cp*IrCl2]2 в присутствии AgOTf дает с выходом 90% продукт C–H- и двойной B–H-активации – катионный комплекс [(Cp*Ir)3(μ-Cl)2(1-(4'-MeOC6H3N=N)-1,12-C2B10H9-κ5-N(2),B(2);N(1), C(2');B(3))]OTf (рис. 31, схема 25 ) [75].
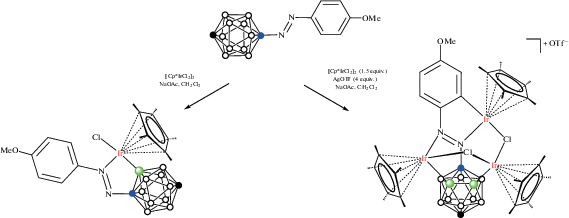
Схема 25.
Рис. 31.
Строение комплексов [Cp*IrCl(1-(4'-MeOC6H4N=N)-1,12-C2B10H10-κ2-N(2),B(2))] (а) и [(Cp*Ir)3(μ-Cl)2(1-(4'-MeOC6H3N=N)-1,12-C2B10H9-κ5-N(2),B(2);N(1),C(2');B(3))]+ (б). Атомы водорода органических заместителей и лигандов не показаны для ясности.
Реакция 1,12-бис(арилазо)производного пара-карборана 1,12-(4'-MeOC6H4N=N)2-1,12-C2B10H10 с 1 экв. [Cp*IrCl2]2 и NaOAc в хлористом метилене дает с выходом 70% синий комплекс [Cp*IrCl(1-(4'-MeOC6H4N=N)2-1,12-C2B10H9-κ2-N(2),B(2))], а реакция с 3 экв. [Cp*IrCl2]2 и 8 экв. AgOTf приводит с выходом 82% к темно-коричневому четырехъядерному катионному комплексу [(Cp*Ir)4(μ-Cl)2Cl(1,12-(4'-MeOC6H3N=N)2-1,12-C2B10H8-κ7-N(2),B(2);N(1),C(2');B(3);N(1'),C(2"))] OTf – продукту C–H- и двойной B–H-активации по одной арилазогруппе и C–H-активации по второй (рис. 32). Последний также может быть получен обработкой синего комплекса 2.5 экв. [Cp*IrCl2]2 и 7 экв. AgOTf (схема 26 ) [75].
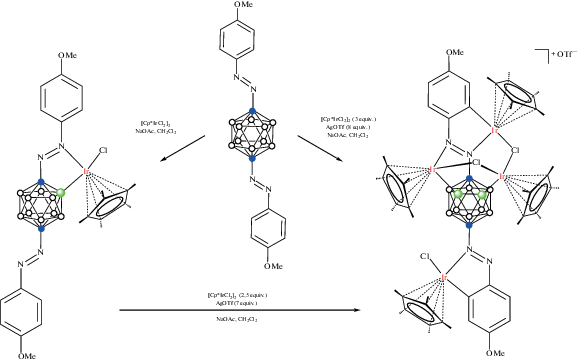
Схема 26.
Рис. 32.
Строение комплексов [Cp*IrCl(1,12-(4'-MeOC6H4N=N)2-1,12-C2B10H9-κ2-N(2),B(2))] (а) и [(Cp*Ir)4(μ-Cl)2Cl(1,12-(4'-MeOC6H3N=N)2-1,12-C2B10H8-κ7-N(2),B(2);N(1),C(2’);B(3);N(1'),C(2"))]+ (б). Атомы водорода органических заместителей и лигандов не показаны для ясности.
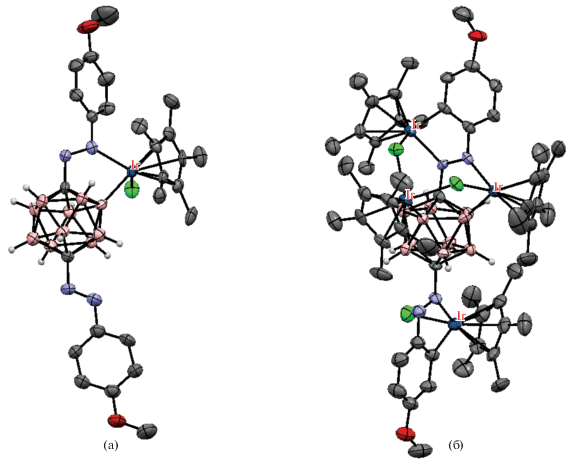
В качестве направляющего лиганда также может выступать пиридильная группа. Так, реакция 1-(2'-пиридил)-2-метил-орто-карборана 1-(2'-NC5H4)-2-Me-1,2-C2B10H10 с ароматическими N-ацилглутаримидами ArC(O)N(CO)2(CH2)3 в присутствии 5 мол. % [(COD)RhCl2]2 в толуоле при 150°С приводит к образованию соответствующих B(3),B(5)-диарилпроизводных 1-(2-NC5H4)-2-Me-3,5-Ar2-1,2-C2B10H8. Аналогичная реакция с третичными алифатическими N-ацилглутаримидами MeR2C(O)N(CO)2(CH2)3 протекает с перегруппировкой алкильного заместителя и приводит к продуктам B(4)-алкилирования 1-(2'-NC5H4)-2-Me-4-R2CHCH2-1,2-C2B10H9 (схема 27 ). Можно предположить, что в первом случае образованию B(3),B(6)-диарилпроизводных препятствует невозможность необходимого поворота направляющей группы после введения первого заместителя в положение 3, а во втором случае замещению в положение 3 препятствует большой объем третичной алкильной группы в сочетании с метильной группой в положении 2 карборанового остова [77].
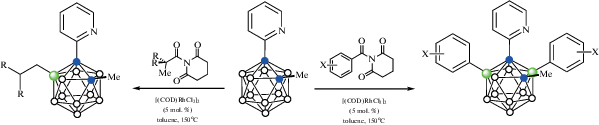
Схема 27.
Реакция 1-(2'-пиридил)-орто-карборана 1-(2'-NC5H4)-1,2-C2B10H11 с арилбороновыми кислотами ArB(OH)2 в присутствии Ag2O и 10 мол. % Pd(OAc)2 в диметилформамиде при комнатной температуре приводит к образованию соответствующих 3-арилпроизводных 1-(2-NC5H4)-3-Ar-1,2-C2B10H10 (схема 28 ). Реакция хорошо протекает с бороновыми кислотами, содержащими как электроноакцепторные (Ar = C6H4-4-F, C6H4-3-F, C6H3-3,5-F2, C6H4-4-Cl, C6H4-4-Br, C6H4-4-CF3, C6H4-3-NO2, C6H4-4-CHO, C6H4-4-COMe, C6H4-4-COOMe, C6H4-4-CN), так и электронодонорные (C6H4-4-Me, C6H4-3-Me, C6H4-2-Me, C6H3-3,4-Me2, C6H4-4-Et, C6H4-4-i-Pr, C6H4-4-t-Bu, C6H4-4-OCH2Ph) заместители, а также с 4-бифенил-, 2-нафтил- и 1-пиренилбороновыми кислотами. Показано, что замещению в положение 3 также не препятствует наличие n-Bu- и Ph-заместителей при соседнем атоме углерода. Этот подход также может быть использован для последовательного введения арильных групп с различными заместителями в положения 3 и 6 орто-карборанового остова [78].
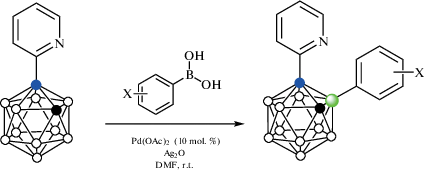
Схема 28.
Реакция 1-(2'-пиридил)-2-метил-орто-карборана 1-(2'-NC5H4)-2-Me-1,2-C2B10H10 с карбоновыми кислотами RCOOH в присутствии Cu(OH)2, оксона, 5 мол. % [Cp*RhCl2]2 и 20 мол. % AgOAc в 1,2-дихлорэтане при 120°С приводит к образованию соответствующих B(3)-ацилоксипроизводных 1-(2-NC5H4)-2-Me-3-RCOO-1,2-C2B10H9 и B(3,6)-диацилоксипроизводных 1-(2-NC5H4)-2-Me-3,6-(RCOO)2-1,2-C2B10H9 (схема 29 ). В качестве кислот могут выступать как алифатические (первичные, вторичные и третичные), так и ароматические и гетероароматические кислоты. Аналогичные реакции протекают также с 1-(2'-пиридил)карборанами, как содержащими при втором атоме углерода другие заместители (например, бензильную группу), так и не содержащими их вовсе, а в качестве направляющей группы может выступать 2-пириримидил [79].
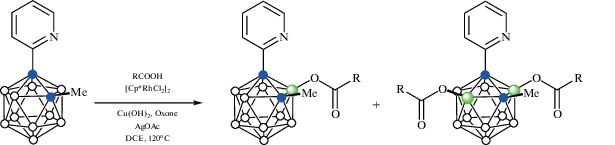
Схема 29.
Следует отметить, что 2-пиридильная группа также может использоваться для направленного селективного окислительного сочетания двух карборановых остовов. Так, реакция 1-(2'-пиридил)-орто-карборана 1-(2'-NC5H4)-1,2-C2B10H11 с AgNO3 и 10 мол. % [(MeCN)4Pd](BF4)2 в толуоле при 60°С приводит к B(3)–H-активации орто-карборана с образованием смеси диастереомерных 3,3'- и 3,6'-бис(1-(2"-пиридил)-орто-карборанов), которые могут быть разделены колоночной хроматографией [80].
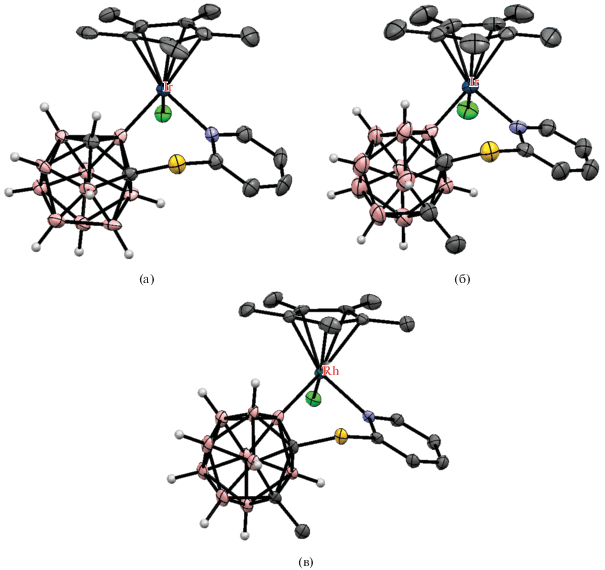
Реакция 1-(2'-пиридилсульфенил)-орто-карборана 1-(2'-NC5H4S)-1,2-C2B10H11 с [Cp*IrCl2]2 в хлористом метилене в присутствии KOAc и AgOTf приводит к образованию B(3)-циклометаллированного комплекса [Cp*IrCl(1-(2'-NC5H4S)-1,2-C2B10H10-κ2-N,B(3))] (рис. 33) с выходом 69%, тогда как аналогичная реакция с 1-(2'-пиридилсульфенил)-2-метил-орто-карбораном 1-(2'-NC5H4S)-2-Me-1,2-C2B10H10, в котором связанный со вторым атомом углерода атом водорода заменен на метильную группу, приводит с выходом 73% к образованию B(4)-циклометаллированного комплекса [Cp*IrCl(1-(2'-NC5H4S)-2-Me-1,2-C2B10H9-κ2-N,B(4))] (рис. 33), это связано с пространственными затруднениями атаки по положению 3, вызванными близлежащей метильной группой (схема 30 ). Интересно, что реакция 1-(2'-NC5H4S)-2-Me-1,2-C2B10H10 с [Cp*RhCl2]2 в аналогичных условиях также дает продукт B(4)-циклометаллирования [Cp*RhCl(1-(2'-NC5H4S)-2-Me-1,2-C2B10H9-κ2-N,B(4))] (рис. 33) (выход 73%), в то время как реакция с не содержащим метильной группы 1-(2'-NC5H4S)-1,2-C2B10H11 приводит к внедрению металла в карборановый остов с образованием родакарборана [3-Cp*-2-(2'-NC5H4)-3,1,2-RhC2B9H10] с выходом 39% (схема 30 ). Необходимо отметить, что в данном случае замещению подвергается атом бора в положении 3 [81].
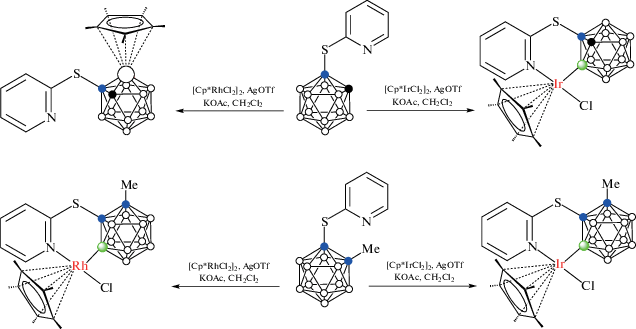
Схема 30.
Рис. 33.
Строение комплексов [Cp*IrCl(1-(2'-NC5H4S)-1,2-C2B10H10-κ2-N,B(3))] (а), [Cp*IrCl(1-(2'-NC5H4S)-2-Me-1,2-C2B10H9-κ2-N,B(4))] (б) и [Cp*RhCl(1-(2'-NC5H4S)-2-Me-1,2-C2B10H9-κ2-N,B(4))] (в). Атомы водорода органических заместителей и лигандов не показаны для ясности.
Обработка родиевого комплекса [Cp*RhCl(1-(2'-NC5H4S)-2-Me-1,2-C2B10H9-κ2-N,B(4))] N-бром- и N-иодсукцинимидами в 1,2-дихлорэтане при 65°C приводит к разрыву связи B–Rh с образованием соответствующих B(4)-галогенопроизводных 1-(2'-NC5H4S)-2-Me-4-X-1,2-C2B10H9 (X = Br, I) с практически количественным выходом, а при его взаимодействии с кислородом воздуха в дихлорметане образуется соответствующее гидроксипроизводное 1-(2'-NC5H4S)-2-Me-4-HO-1,2-C2B10H9. Следует отметить, что продукты замещения в положение 4 карборанового остова 1-(2'-NC5H4S)-2-R-4-X-1,2-C2B10H9 (R = Me, Bn, SiMe3; X = Br, OH) могут быть получены при проведении реакции в присутствии [Cp*RhCl2]2 (но не [Cp*IrCl2]2) без выделения промежуточного металлокомплекса [81].
Еще одним типом направляющего лиганда может служить 1,2,3-триазольная группа. Реакция 1-(1'-триазолметил)-орто-карборанов 1-(4'-R-1',2',3'-N3C2H-1'-CH2)-1,2-C2B10H11 (R = Ph, Bu) с [Cp*IrCl2]2 в хлористом метилене в присутствии NaOAc при комнатной температуре приводит к образованию соответствующих B(3)-циклометаллированных комплексов [Cp*IrCl(1-(4'-R-1',2',3'-N3C2H-1'-CH2)-1,2-C2B10H10-κ2-N(2),B(3))] (рис. 34) с выходом 74 и 52%. В случае карборанилтриазолов, содержащих металлоценовые заместители (ферроцен или рутеноцен), реакция требует более жестких условий – присутствия Cs2CO3 как основания и нагревания в ацетонитриле, приводя к соответствующим циклометаллированным комплексам с выходом 44–46% (схема 31 , рис. 34). Аналогичные реакции с [Cp*RhCl2]2 приводят к циклометаллированным комплексам с металлоценовыми заместителями с выходом 35–46% (схема 31 ), в то время как комплексы с фенильным и бутильным заместителями получить не удалось [82].
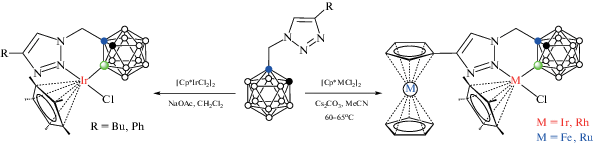
Схема 31.
Рис. 34.
Строение комплекса [Cp*IrCl(1-(4'-Fc-1',2',3'-N3C2H-1'-CH2)-1,2-C2B10H10-κ2-N(2),B(3))] (атомы водорода органических заместителей и лигандов не показаны для ясности).
В свою очередь, обработка карборанилтриазолиевых солей [1-(3'-Me-4'-R-1',2',3'-N3C2H-1'-CH2)-1,2-C2B10H11]+[BF4]– (R = Ph, Bu) с Ag2O/Me4NCl в дихлорметане при комнатной температуре с последующим добавлением [Cp*MCl2]2 (M = Ir, Rh) приводит к образованию соответствующих C(2)-циклометаллированных карбеновых комплексов [Cp*IrCl(1-(3'-Me-4'-R-1',2',3'-N3C2H-1'-CH2)-1,2-C2B10H10-κ2-C(2),C(5'))] (схема 32 ). Следует отметить, что C(2)-циклометаллированные комплексы могут быть получены при депротонировании незамещенной CH-группы карборана. Так, обработка 1-(4'-Fc-1',2',3'-N3C2H-1'-CH2)-1,2-C2B10H11n-BuLi в тетрагидрофуране с последующим добавлением [Cp*IrCl2]2 приводит к [Cp*IrCl(1-(4'-Fc-1',2',3'-N3C2H-1'-CH2)-1,2-C2B10H10-κ2-N(2),C(2))] (схема 32 ) [82].
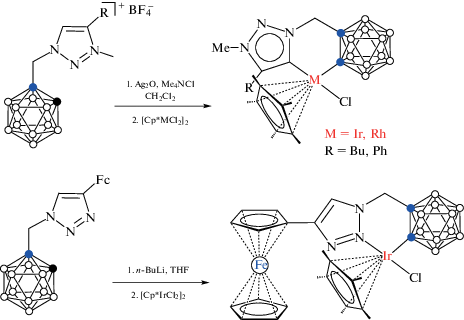
Схема 32.
В качестве переходного металла в процессе каталитической B–H-активации орто-карборана также могут использоваться соли меди, однако при этом требуется окисление меди в каталитическом цикле. Так, взаимодействие 1-(8'-хинолиламинокарбонил)-орто-карборана 1-(8'-NC9H6NH(O)C)-2-R-1,2-C2B10H10 c терминальными арилацетиленами в присутствии Cu(OTf)2, 2-фенилпиридина, Ag2CO3 и K2HPO4 в 1,2-дихлорэтане при 130°С с последующей обработкой HCl в хлороформе приводит к соответствующим 4-арилацетиленам 1-(8'-NC9H6NH(O)C)-4-ArC≡C-2-R-1,2-C2B10H9 (схема 33 ). Роль окислителя в этом процессе играет Ag2CO3. Реакция протекает через образование ацетиленовых комплексов меди(I) [Cu(1-(8-NC9H6N(O)C)-4-ArC≡C-2-R-1,2-C2B10H9-η2-C,C-κ2-N,N))], строение одного из которых было установлено методом рентгеноструктурного анализа (рис. 35) [83].
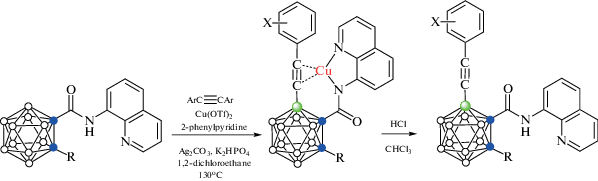
Схема 33.
Рис. 35.
Строение комплекса [Cu(1-(8'-NC9H6N(O)C)-4-(4'-BrC6H4C≡C-2-R-1,2-C2B10H9-η2-C,C-κ2-N,N))] (атомы водорода не показаны для ясности).
Реакция амидов орто-карборанилкарбоновых кислот и 8-аминохинолина 1-(8'-NC9H6NH(O)C)-2-R-1,2-C2B10H10 (R = Me, Et, Pr, i-Pr, Bu, Ph) с t-BuOLi в присутствии 20 мол. % Cu(OTf)2 при 4.0 мА приводит к соответствующим 4-трет-бутоксипроизводным, в то время как реакция с 1-(2'-NC5H4CMe2NH(O)C)-2-Me-1,2-C2B10H10 в аналогичных условиях приводит к соответствующему 4,5-ди(трет-бутокси)производному. Гидролиз полученных трет-бутоксипроизводных в 12 M соляной кислоте при 120°C приводит к соответствующим гидроксипроизводным, а нагревание с K2CO3 в тетрагидрофуране – к гидролизу амида и декарбоксилированию, т.е. к полному удалению направляющего лиганда. Реакция 1-(8'-NC9H6NH(O)C)-2-i-Pr-1,2-C2B10H10 c различными фенолятами лития ArOLi в аналогичных условиях приводит к соответствующим 4,5-ди(фенокси)производным 1-(8'-NC9H6NH(O)C)-4,5-(ArO)2-2-i-Pr-1,2-C2B10H8 [84].
Взаимодействие 1-(8'-NC9H6NH(O)C)-2-Me-1,2-C2B10H10 c избытком различных диарил- и алкилдисульфидов RSSR в присутствии Cu(OTf)2, 2-фенилпиридина и t-BuOLi в 1,2-дихлорэтане при 130°C приводит к соответствующим 7,11-дисульфенилпроизводным 7,11-(RS)2-1-Me-1,2-C2B10H9. Реакция протекает через B–H-активацию положений 4 и 5, которые после удаления направляющего лиганда в форме N,N'-ди(8-хинолил)мочевины превращаются в положения 7 и 11. Реакция толерантна к различным функциональным группам, включая -F, -Cl, -Br, -CF3, -OMe и ‑SMe. Метильная группа при втором атоме углерода может быть заменена на i-Pr, Bu-, Ph, CH2C6H4-4-Me. Предполагается, что роль окислителя в данной реакции играет избыток дисульфида [85].
Реакции B–H-активации с участием кислородсодержащих направляющих лигандов
Для направленного введения разнообразных заместителей в карборановый остов с помощью направляющих донорных групп наибольшее внимание получили производные с различными кислородсодержащими заместителями – в первую очередь карбоновыми кислотами и их амидами, что во многом связано с низкой устойчивостью образующихся промежуточных комплексов переходных металлов со связью металл–кислород, а также возможностью легкого удаления направляющей карбоксильной группы с помощью термического декарбоксилирования, которое значительно облегчается наличием заместителей в соседних положениях остова. Следует отметить, что во всех случаях (даже в отсутствие заместителя при соседнем атоме углерода карборана) карбоксильная группа способствует B–H-активации положений 4 и 5, которые при наличии дополнительного заместителя при втором атоме углерода после удаления карбоксильной группы становятся положениями 7 и 11 соответственно.
Первым примером использования реакции B–H-активации для синтеза различных производных орто-карборана была реакция орто-карборанилкарбоновой кислоты 1-HOOC-1,2-C2B10H11 с диарилацетиленами Ar-C≡C-Ar в присутствии Cu(OAc)2, AgOAc и 2.5 мол. % [Cp*IrCl2]2 в толуоле при 180°C, протекающая с потерей направляющей группы и приводящая к соответствующим B(4)-замещенным алкенам 4-ArHC=C(Ar)‑1,2‑C2B10H11 (схема 34 ). Замещение в положение 4 в данном случае обусловлено чисто электронными факторами. Аналогичные реакции с C,C'-замещенными кислотами 1-HOOC-2-R-1,2-C2B10H10 (R = Me, Bn) приводят к соответствующим B(7)-замещенным производным 7-PhHC=C(Ph)-1-R-1,2-C2B10H10. Предполагается, что реакция протекает через образование циклометаллированного комплекса, аналог которого [Cp*Ir(1-OOC-2-Me-1,2-C2B10H9-κ2-O,B(4))(DMSO-κ1-S)] (рис. 36) был получен реакцией 1-HOOC-2-Me-1,2-C2B10H10 с [Cp*Ir(OAc)2(DMSO)] в толуоле [18].
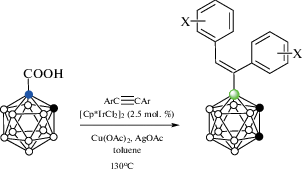
Схема 34.
Рис. 36.
Строение комплекса [Cp*Ir(1-OOC-2-Me-1,2-C2B10H9-κ2-O,B(4))(DMSO-κ1-S)] (атомы водорода органических заместителей и лигандов не показаны для ясности).
Реакция орто-карборанилкарбоновой кислоты 1-HOOC-2-Me-1,2-C2B10H10 с терминальными ацетиленами Ar-C≡CH и R3Si-C≡CH в присутствии AgOAc, K2HPO4 и 5 мол. % Pd(OAc)2 в толуоле при 80°C приводит к соответствующим B(7)-замещенным ацетиленам 7-ArC≡C-1-Me-1,2-C2B10H10 и 7-R3SiC≡C-1-Me-1,2-C2B10H10 соответственно с выходом, варьирующимся от 44 до 86% (схема 35 ). Замена в орто-карборанилкарбоновой кислоте метильной группы при соседнем атоме углерода на другие алкильные группы (i-Pr, Bn) практически не сказывается на выходе продуктов алкинилирования, однако в отсутствие заместителя выход ацетилена 4-i-Pr3SiC≡C-1,2-C2B10H11 падает до 35% (при использовании в качестве исходного соединения 1-HOOC-2-Me3Si-1,2-C2B10H10 выход 4-i-Pr3SiC≡C-1,2-C2B10H11 составляет 74%). Удаление силильной защиты обработкой фторид-ионом приводит к терминальным карборанилацетиленам, которые способны вступать в различные реакции, характерные для других терминальных ацетиленов, такие как реакция [3+2]-циклоприсоединения азидов к ацетиленам (“клик-реакция”), реакции Соногаширы и Глазера–Хея [86].
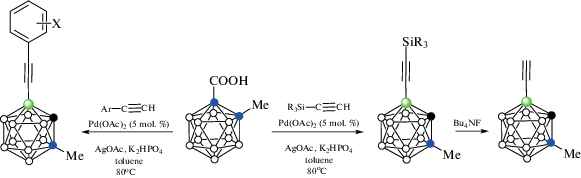
Схема 35.
Реакция орто-карборанилкарбоновой кислоты 1-HOOC-1,2-C2B10H11 со стиролом в присутствии AgOAc и 10 мол. % Pd(OAc)2 в 1,2-дихлорэтане при 60°C приводит к 4,5-диалкенпроизводному 4,5-(PhHC=CH)2-1,2-C2B10H10. Ряд 7,11-диалкенпроизводных 7,11-(PhHC=CH)2-1-R-1,2-C2B10H9 (R = = Me, Et, i-Pr, Bn, Ph, C6H4-4-Cl, C6H3-3,5-Me2, SiMe3) был получен взаимодействием орто-карборанилкарбоновых кислот 1-HOOC-2-R-1,2-C2B10H10 с замещенными стиролами в аналогичных условиях (схема 36 ) [87].
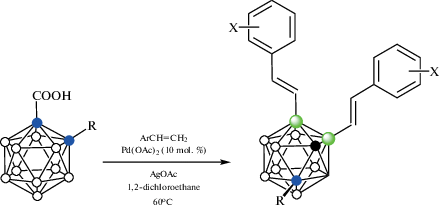
Схема 36.
Взаимодействие орто-карборанилкарбоновой кислоты 1-HOOC-2-Me-1,2-C2B10H10 с арилиодидами в присутствии AgOAc, уксусной кислоты и 10 мол. % Pd(OAc)2 в толуоле при 70°C приводит к соответствующим 7,11-диарилпроизводным 7,11-Ar2-1-Me-1,2-C2B10H9 (схема 37 ). В реакцию вступают арилиодиды, содержащие как электронодонорные (3-Me, 4-Me, 3,5-Me2, 4-t-Bu, 4-OMe), так и электроноакцепторные (4-Ph, 3-Cl, 4-Cl, 3-Br, 4-Br, 4-F, 4-COMe, 3-COOMe) заместители, при этом выход диарилпроизводных варьируется от 50 до 82%. Метильная группа в орто-карборанилкарбоновой кислоте может быть заменена на фенильную или бензильную, в то время как в случае 1-HOOC-1,2-C2B10H11 реакция приводит к 4,5-Ph2-1,2-C2B10H10 с выходом 41% (использование Me3Si-группы приводит к повышению выхода до 69%) [88].
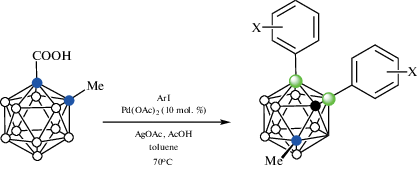
Схема 37.
Взаимодействие 1-HOOC-2-R-1,2-C2B10H10 с тиофеном в присутствии Li2CO3, Cu(OAc)2, 5 мол. % [Cp*IrCl2]2, 10 мол. % AgNTf2 и 10 мол. % AgOAc в толуоле при 130°C приводит к соответствующим 7-(2'-тиенил)производным орто-карборана. Реакция протекает с хорошим выходом с различными замещенными производными тиофена, бензотиофенами, тиенотиофенами, а также олиготиофенами [89].
Реакция орто-карборанилкарбоновой кислоты 1-HOOC-2-Hx-1,2-C2B10H10 с аллильными спиртами HOCH(R)CH=CH2 (R = Alk, Ar) в присутствии AgOAc и 5 мол. % [Cp*RhCl2]2 в 1,4-диоксане при 70°C приводит к соответствующим 7,11-диалкилпроизводным 7,11-(RC(O)CH2CH2)2-1-Hx-1,2-C2B10H9 (схема 38 ). Гексильная группа в орто-карборанилкарбоновой кислоте может быть заменена бутильной или фенильной, однако в отсутствие заместителя реакция приводит лишь к следовым количествам целевого продукта [90].
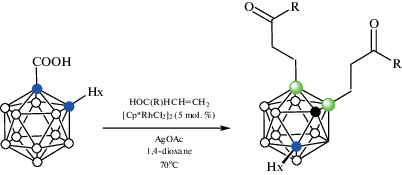
Схема 38.
Взаимодействие орто-карборанилкарбоновой кислоты 1-HOOC-2-Me-1,2-C2B10H10 c замещенными пропаргиловыми спиртами Ar-C≡C-CR1R2OH в присутствии NaOAc и каталитических количеств [Cp*IrCl2]2 и AgSbF6 в трифторэтаноле при 60°C протекает через образование B(4)-алкенильных производных, которые претерпевают циклизацию в соответствующие индены, а последующее декарбоксилирование в толуоле при 130°C приводит к удалению направляющей группы (схема 39 ). Метильная группа в орто-карборанилкарбоновой кислоте может быть заменена бутильной или арильной [91].
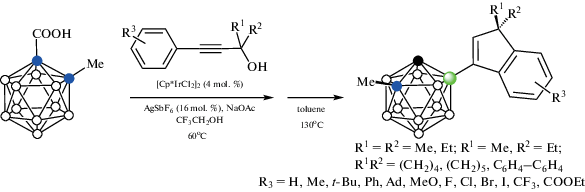
Схема 39.
Интересно, что реакция в мезитилене протекает без циклизации и приводит к соответствующим диеновым производным (схема 40 ) [91].
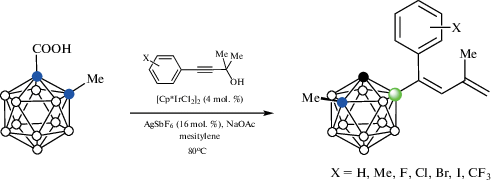
Схема 40.
Реакция орто-карборанилкарбоновых кислот 1-HOOC-2-R-1,2-C2B10H10 (R = H, Me, Et, i-Pr, Bu) с 2-фенилпиридином в присутствии каталитических количеств [Cp*IrCl2]2, AgOTf, AgOAc и избытка Cu(OAc)2 в толуоле при 130°C приводит к арилпроизводным, в которых пиридиновый фрагмент играет роль направляющей группы по отношению к замещению в бензольном кольце. Замещение протекает в положение 4 по отношению к направляющей карбоксильной группе, однако с учетом декарбоксилирования арильный заместитель оказывается в положении 7 по отношению к заместителю при втором атоме углерода 7-(2"-NC5H4-2'-C6H4)-1-R-1,2-C2B10H10 (схема 41 ). Реакции c производными С-метил, С-этил и С-бутил протекают с высоким выходом (73–93%), в случае же материнской орто-карборанилкарбоновой кислоты 1-HOOC-1,2-C2B10H11 и производного С-изопропил выход составляет 51 и 41% соответственно. Реакция толерантна к широкому кругу заместителей как в бензольном, так и в пиридиновом кольце (F, Cl, Br, Me, t-Bu, CF3, OMe, CHO, CN), а также замене пиридина другими азотсодержащими направляющими группами, такими как хинолин, пиримидин, тиазол, пиразол, индол, -C(Me)=NOMe, -N(O)=NPh или ‑C(OEt)=NH. В последнем случае направляющая группа теряет молекулу этанола, приводя к образованию 7-(2'-цианофенильного) производного 7-(2'-NCC6H4)-1-Me-1,2-C2B10H10. Реакция протекает через образование циклометаллированного комплекса иридия(III) с 2-фенилпиридином и орто-карборанилкарбоксилатом (рис. 37), окисление которого Cu(OAc)2 в присутствии AgOTf приводит к образованию борметалированного комплекса с последующим внедрением лиганда по связи бор–металл. Реакция также применима к гетероароматическим соединениям (тиофен, пиррол, индол) с азотсодержащими направляющими группами [92].
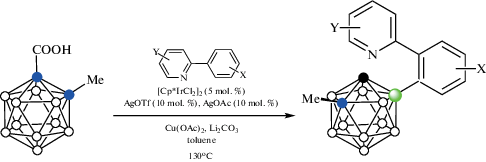
Схема 41.
Рис. 37.
Строение комплекса [Cp*Ir{2'-(2"-NC5H4)C6H3-5'-CF3-κ2-N,C(2')}(1-OOC-2-Me-1,2-C2B10H10-κ1-O)] (атомы водорода органических лигандов не показаны для ясности).
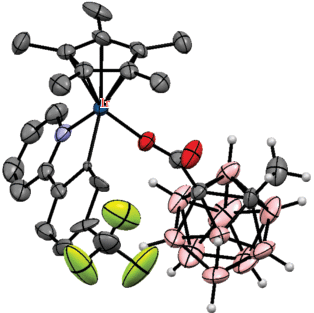
Рис. 38.
Строение комплекса [(MeCN)2Pd(1-OOC-1-CB11H10-κ2-O,B(2))]– (атомы водорода органических лигандов не показаны для ясности).
Реакция орто-карборанилкарбоновой кислоты 1-HOOC-2-Me-1,2-C2B10H10 с бензойными кислотами в присутствии каталитических количеств [Cp*IrCl2]2, AgNTf, AgOAc и избытка Li2CO3 и Cu(OPiv)2 в толуоле при 160°C приводит к образованию 4,5-замещенных аналогов кумарина (схема 42 ) [93].
Схема 42.
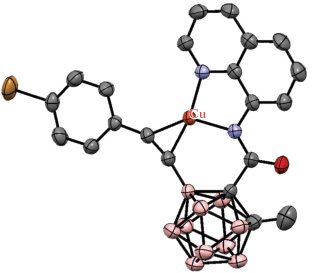
Показано, что карбоксильная группа также может использоваться в качестве направляющего заместителя в синтезе производных карба-клозо-додекаборатного аниона. Реакция 1-карбокси-1-карба-клозо-додекаборана (Et4N)[1-HOOC-1-CB11H11] с избытком арилиодидов в присутствии AgOAc, уксусной кислоты и 2.5 мол. % Pd(OAc)2 в диметилформамиде при комнатной температуре приводит к соответствующим пентаарилпроизводным (Et4N)[1-HOOC-2,3,4,5,6-Ar5-1-CB11H6]. Реакция протекает с высоким выходом (70–94%) при наличии электронодонорных (3-Me, 4-Me, 3,5-Me2, 4-Et, 4-Hx, 4-t-Bu, 4-Bn, 4-OMe, 4-CH2OH, 4-NHAc) и особенно электроноакцепторных (4-Ph, 3-F, 4-F, 3,4-F2, 3-Cl, 4-Cl, 3,5-Cl2, 4-Br, 4-COOMe, 4-COOEt, 4-CHO) заместителей. Проведение реакции при 60°С приводит к декарбоксилированию с образованием (Et4N)[2,3,4,5,6-Ar5-1-CB11H7]. Предполагается, что реакция протекает через образование пятичленного палладацикла, аналогичного комплексу (Me3NH)[(MeCN)2Pd(1-OOC-1-CB11H10-κ2-O,B(2))] (рис. 38 ), который был получен реакцией (Et4N)[1-HOOC-1-CB11H11] с Pd(OAc)2 в ацетонитриле и реагирует с 4-FC6H4I c образованием [2,3,4,5,6-(4'-FC6H4)5-1-CB11H7]– [94].
Реакции 1-карбокси-1-карба-клозо-додекаборана (Et4N)[1-HOOC-1-CB11H11] с избытком замещенных стиролов RCH=CH2 (R = Ph, C6H4-4-F, C6H4-4-CF3, C6H4-4-CN, C6F5), аллилбензолов RCH2CH=CH2 (R = Ph, C6F5) и терминальных алкенов R-CH=CH2 (R = Bu, i-Bu, CH2CHMeEt, CH2CMe3, CH2CH2Ph, CH2CH2COOEt, CH2OPh) в присутствии AgOAc и 10 мол. % Pd(OAc)2 в ацетонитриле при комнатной температуре приводят к соответствующим пентавинильным замещенным (Et4N)[1-HOOC-2,3,4,5,6-(R*CH=CH)5-1-CB11H6]. Производные 1-карба-клозо-додекаборана (Et4N)[1-HOOC-2,3,4,5,6-(ArCH=CH)5-12-X-1-CB11H5] (X = Cl, Ar = Ph; X = Br, Ar = C6H4-4-F; X = Me, Ar = C6H4-4-F; X = Cl, Ar = Ph, C6H4-4-F; X = CN, Ar = C6H4-4-F), содержащие заместители в положении 12, были получены аналогичным образом исходя из соответствующих кислот (Et4N)[1-HOOC-12-X-1-CB11H10]. Восстановление алкенов водородом в присутствии 10% Pd/C приводит к алканам (Et4N)[1-HOOC-2,3,4,5,6-(RCH2CH2)5-1-CB11H6] (R = C6H4-4-F, CH2CMe3). Карбоксильная группа может быть удалена при микроволновом облучении в диметилацетамиде в присутствии NaOAc при 150°C [95].
Карбоксильная группа также может использоваться для получения производных карборана со связью бор–азот. Так, реакция орто-карборанилкарбоновой кислоты 1-HOOC-2-Me-1,2-C2B10H10 с O-бензоилгидроксиламинами PhC(O)ONR2 в присутствии K2HPO4 и 10 мол. % Pd(OAc)2 в толуоле при 100°C приводит к соответствующим 7-замещенным аминам 7-R2N-1-Me-1,2-C2B10H10 (схема 43 ). Восстановление 7-Bn2N-1-Me-1,2-C2B10H10 водородом в растворе HCl в тетрагидрофуране в присутствии 10% Pd/C при комнатной температуре приводит с количественным выходом к амину 7-H2N-1-Me-1,2-C2B10H10 [96].
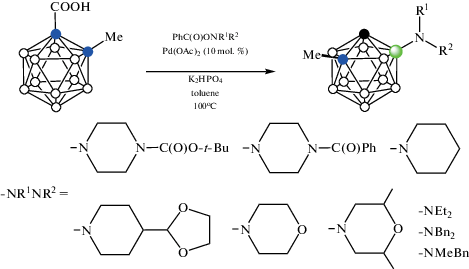
Схема 43.
Реакции орто-карборанилкарбоновых кислот 1-HOOC-2-R-1,2-C2B10H10 (R = Me, Et, i-Pr, CH2Ph, CH2C6H4-4-Me, CH2C6H4-4-Cl, CH2C6H4-4-OMe, CH2CH2OMe, SiMe3) с тозилазидом p-MeC6H4SO2N3 в присутствии NaOAc и 2.5 мол. % [(p-cymene)RuCl2]2 в толуоле при 100°C приводят к соответствующим 7-тозиламинам 7-TsNH-1-R-1,2-C2B10H10 с выходом 84–94% (67% для R = SiMe3) (схема 44 ). Аналогичным образом реакции 1-HOOC-2-Me-1,2-C2B10H10 c PhSO2N3, p-MeOC6H4SO2N3, p-CF3C6H4SO2N3, p‑ClC6H4SO2N3, p-O2NC6H4SO2N3, 1‑NaphSO2N3, BnSO2N3 и BuSO2N3 приводят к соответствующим сульфонамидам [96].
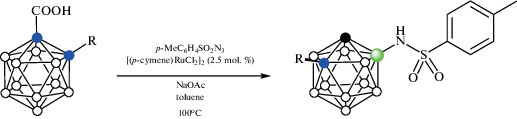
Схема 44.
Реакции 1-HOOC-2-Me-1,2-C2B10H10 с 3-арил- и алкил-1,4,2-диоксазолонами в присутствии NaOAc и каталитических количеств [(p-cymene)RuCl2]2 и AgNTf2 в 1,2-дихлороэтане при 80°C приводят к соответствующим амидам 7-R(O)CNH-1-Me-1,2-C2B10H10 (схема 45 ). Показано, что диоксазолоны являются гораздо более реакционноспособными в реакциях амидирования, чем сульфонилазиды, причем 3-алкил-1,4,2-диоксазолоны более реакционноспособны, чем 3-арил-1,4,2-диоксазолоны, а среди последних несколько более реакционноспособными являются содержащие электроноакцепторные заместители. 2-Фурил- и 2-тиофениламиды, а также 2-нафталил- и 2-азулениламиды были получены аналогично. Метильная группа в орто-карборанилкарбоновой кислоте может быть заменена другими алкильными (бутил, циклогексил) или арильными группами [97].
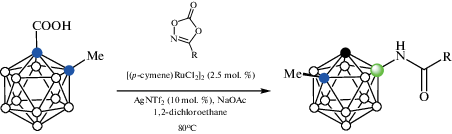
Схема 45.
7-Арил- и алкиламиды 7-R(O)CNH-1-Me-1,2-C2B10H10 также могут быть получены взаимодействием 1-HOOC-2-Me-1,2-C2B10H10 с соответствующими 3-арил- и алкил-1,4,2-диоксазолонами в присутствии NaOAc и каталитических количеств [Cp*IrCl2]2 и AgSbF6 в 1,2-дихлороэтане при комнатной температуре с последующим декарбоксилированием при 80°C (схема 46 ) [98].
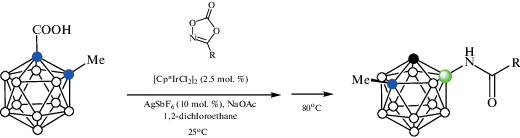
Схема 46.
Карбоксильная группа также может использоваться для введения в положение 4 или 7 (в зависимости от наличия заместителя при втором атоме углерода орто-карборана) простых заместителей, таких как гидроксигруппа или атомы галогена. Так, взаимодействие орто-карборанилкарбоновых кислот 1-HOOC-2-R-1,2-C2B10H10 с кислородом в присутствии KOAc и 2.5 мол. % [Cp*RhCl2]2 в толуоле при 95°C приводит к 7-гидроксипроизводным 7-HO-1-R-1,2-C2B10H10 с выходом 40–80% (схема 47 ). В случае 1-HOOC-2-Me3Si-1,2-C2B10H10 с выходом 30% образуется 4-гидрокси-орто-карборан 4-HO-1,2-C2B10H11 [99]. Взаимодействие 1-HOOC-2-R-1,2-C2B10H10 с различными галогенирующими агентами (FeCl3/Cu(OAc)2, N-бром- и N-иодсукцинимид) в присутствии NaOAc, 2.5 мол. % [Cp*IrCl2]2 и 10 мол. % AgNTf2 в 1,2-дихлорэтане при 100–120°C приводит к соответствующим 7-галогенпроизводным 7-X-1-R-1,2-C2B10H10 (X = Cl, Br, I) с выходом 75–95% (схема 47 ). В случае незамещенной орто-карборанилкарбоновой кислоты 1-HOOC-1,2-C2B10H11 выход 4-иодпроизводного 4-I-1,2-C2B10H11 составил 62%. Реакция 1-HOOC-2-R-1,2-C2B10H10 с IOAc (полученным in situ из I2 и PhI(OAc)2) в присутствии NaOAc и 15 мол. % Pd(OAc)2 в толуоле при 60°C приводит к соответствующим 7,11-дииодпроизводным 7,11-I2-1-R-1,2-C2B10H9 с выходом 68–86% (схема 47 ). Реакция 1-HOOC-1,2-C2B10H11 приводит к 4,5-дииод-орто-карборану 4,5-I2-1,2-C2B10H10 с выходом 62–70% [100].
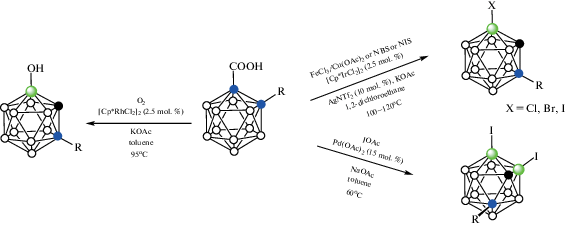
Схема 47.
Отдельный интерес представляют реакции B–H-активации карборанов, содержащих две направляющие карбоксильные группы. Реакция комплекса [(Cp*IrCl2)2(μ-pz)] (pz – пиразин) c орто-карборандикарбоновой кислотой 1,2-(HOOC)2-1,2-C2B10H10 в присутствии AgOTf и Et3N в хлористом метилене при комнатной температуре приводит к B(4,7)–H-активации с образованием тетраядерного металлоцикла {[(Cp*Ir)2(μ-1,2-(OOC)2-1,2-C2B10H8-κ4-O,B(4);O,B(7))](μ-pz)}2 (схема 48 , рис. 39). Интересно, что аналогичная реакция с орто-карборанилкарбоновой кислотой 1-HOOC-1,2-C2B10H11 приводит к продукту C–H-активации – двухъядерному комплексу [(Cp*Ir(1-OOC-1,2-C2B10H10-κ2-O,С))2(μ-pz)] (рис. 40) [101].
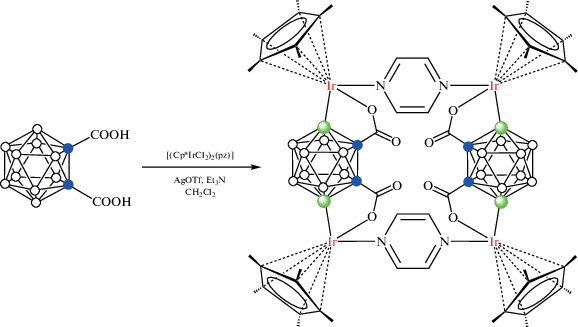
Схема 48.
Рис. 39.
Строение комплекса {[(Cp*Ir)2(μ-1,2-(OOC)2-1,2-C2B10H8-κ4-O,B(4),O,B(7))](μ-pz)}2 (атомы водорода органических лигандов не показаны для ясности).
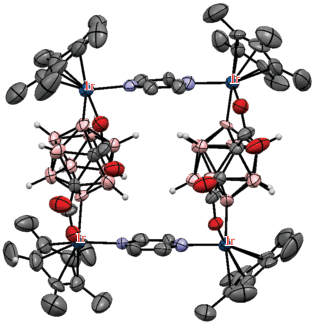
Рис. 40.
Строение комплексов [(Cp*Ir(1-OOC-1,2-C2B10H10-κ2-O,С))2(μ-pz)] (а) и [(Cp*Ir)2(μ-Cl)(μ-1,7-(OOC)2-1,7-C2B10H8-κ4-O,B(2);O,B(3))]– (б). Атомы водорода органических лигандов не показаны для ясности.
Реакция комплекса [(Cp*IrCl2)2(μ-pz)] c мета-карборандикарбоновой кислотой 1,7-(HOOC)2-1,7-C2B10H10 в присутствии AgOTf и Et3N в хлористом метилене при комнатной температуре приводит к B(2,3)–H-активации с образованием двухъядерного комплекса (Et3NH)[(Cp*Ir)2(μ-Cl)(μ-1,7-(OOC)2-1,7-C2B10H8-κ4-O,B(2);O,B(3))] (рис. 40). Реакция комплекса [(Cp*IrCl2)2(μ-pz)] c пара-карборандикарбоновой кислотой 1,12-(HOOC)2-1,12-C2B10H10 в аналогичных условиях приводит к B(2,9)–H-активации с образованием тетраядерного металлоцикла {[(Cp*Ir)2(μ-1,12-(OOC)2-1,12-C2B10H8-κ4-O,B(2);O,B(9))](μ-pz)}2 (рис. 41) [101].
Рис. 41.
Строение комплекса {[(Cp*Ir)2(μ-1,12-(OOC)2-1,12-C2B10H8-κ4-O,B(2);O,B(9))](μ-pz)}2 (атомы водорода органических лигандов не показаны для ясности).
В качестве направляющего лиганда может также быть амидная группа. Так, реакция орто-карбораниламидов 1-R2N(O)C-2-Ph-1,2-C2B10H10 (R2 = Me2, Et2, i-Pr2, (CH2)4, (CH2CH2)2O) с различными арилиодидами в присутствии каталитических количеств Pd(OAc)2 и Ag2CO3 в гексафторизопропаноле при комнатной температуре приводит к соответствующим 3-арилпроизводным 1-R2N(O)C-3-Ar-2-Ph-1,2-C2B10H9, в то время как аналогичная реакция в присутствии Pd(OAc)2 и Ag(CF3COO) приводит к продуктам 3,4-диарилирования 1-R2N(O)C-3,4-Ar-2-Ph-1,2-C2B10H8 (схема 49 ). Реакции толерантны к различным функциональным группам, включая хлор, бром, метил, формил, ацетил и нитрогруппы, причем выход обеих реакций арилирования практически не зависит от электронного эффекта заместителя в бензольном кольце [102].
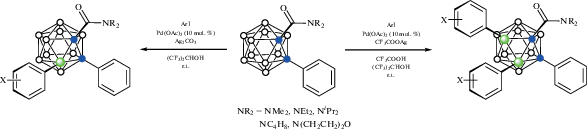
Схема 49.
Амидная группа также была использована в качестве направляющего заместителя в синтезе производных 1-карба-клозо-декаборатного аниона. Так, взаимодействие 1-изопропиламинкарбонил-1-карба-клозо-декаборана (Et4N)[1-i-PrHN(O)C-1-CB9H9] с избытком алкил-, бензил- и арилакрилатов, винилсульфонами и стиролами в присутствии Cu(OAc)2 ‧ H2O, 40 мол. % AgSbF6 и 10 мол. % [Cp*RhCl2]2 в N,N-диметилацетамиде при 20–45°C приводит к соответствующим 2,3-дивинильным производным (Et4N)[1-iPrHN(O)C-2,3-(XCH = = CH)5-1-CB9H7] (X = COOR (R = Alk, Bn, Ar), SO2R (R = Me, Ph), Ar) (схема 50 ) [103].
Схема 50.
В качестве направляющей группы для модификации карборанов также может рассматриваться фосфиноксидная группа. Взаимодействие орто-карборанилфосфиноксидов 1-Ph2P(O)-2-Ar-1,2-C2B10H10 с арилбороновыми кислотами Ar'B(OH)2 в присутствии перекиси водорода, трифторуксусной кислоты и 10 мол. % Pd(OAc)2 в толуоле при 40°C приводит с умеренным выходом (44–73%) к соответствующим 3-арилпроизводным 1-Ph2P(O)-3-Ar'-2-Ar-1,2-C2B10H9 (схема 51 ). Реакция 1-Ph2P(O)-1,2-C2B10H11 с 4-МеC6H4B(OH)2 дает 1-Ph2P(O)-3-(4'-MeC6H4)-1,2-C2B10H10 с выходом 43% [104].
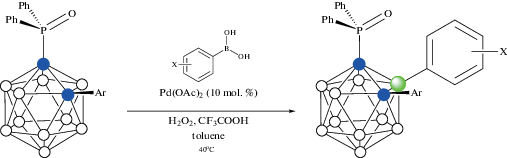
Схема 51.
Еще одним типом направляющих групп с кислородным донорным атомом могут служить аминоацильные группы, которые, в отличие от карбониламидных групп, связаны не с атомами углерода, а с атомами бора. Так, реакция легкодоступных 3-ацетиламино-орто-карборанов 3-MeC(O)NH-1,2-R2-1,2-C2B10H9 (R = H, Me) с арилиодидами в присутствии Ag2CO3, Cu(OTf)2 и 5 мол. % Pd(OAc)2 в 1,2-дихлорэтане при 60°C приводит к соответствующим 8-арилпроизводным 3-MeC(O)NH-8-Ar-1,2-R2-1,2-C2B10H8 (R = = H, Me) (схема 52 ). Выход целевых продуктов слабо зависит от наличия в ароматическом кольце электронодонорных или электроноакцепторных заместителей и находится в пределах 62–85%. В качестве направляющей группы может использоваться 3-бензоиламин 3-PhC(O)NH-, а также его замещенные производные. Интересно, что в выделенном в присутствии 1,10-фенантролина промежуточном циклометаллированном комплексе [(Phen)Pd(3-MeC(O)NH-1,2-Me2-1,2-C2B10H8-κ2-O,B(4))]+[OTf]– атом палладия связан не с атомом бора в положении 8, а с атомом бора в соседнем положении 4 (рис. 42). По-видимому, это связано с миграцией атома палладия на следующей стадии реакции. По данным квантово-химических расчетов, геометрия B(4)-металлированного комплекса [(Phen)Pd(II)(3-MeC(O)NH-1,2-Me2-1,2-C2B10H8-κ2-O,B(4))]+ на 0.7 ккал/моль выгоднее, чем B(8)-металлированного аналога [(Phen)Pd(II)(3-MeC(O)NH-1,2-Me2-1,2-C2B10H8-κ2-O,B(8))]+, однако после присоединения иодбензола геометрия B(8)-металлированного комплекса [(Phen)(Ph)IPd(IV)(3-MeC(O)NH-1,2-Me2-1,2-C2B10H8-κ2-O,B(8))]+, напротив, становится на 5.1 ккал/моль более выгодной, чем соответствующего B(4)-металлированного комплекса. Следует отметить, что реакция B(4)-металлированного комплекса [(Phen)Pd(3-MeC(O)NH-1,2-Me2-1,2-C2B10H8-κ2-O,B(4))]+[OTf]– с иодбензолом приводит к продукту B(8)-арилирования 3-MeC(O)NH-8-Ph-1,2-Me2-1,2-C2B10H8 [105].
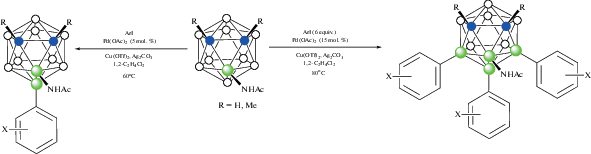
Схема 52.
Рис. 42.
Строение комплекса [(Bipy)Pd(3-MeC(O)NH-1,2-Me2-1,2-C2B10H8-κ2-O,B(4))]+ (атомы водорода органических заместителей и лигандов не показаны для ясности).
Реакция 3-ациламино-орто-карборанов 3-RC(O)NH-1,2-C2B10H11 (R = Me, Ph) с избытком арилиодидов в аналогичных условиях приводит к соответствующим 4,7,8-триарилпроизводным 3-RC(O)NH-4,7,8-Ar3-1,2-C2B10H8 (схема 52 ). Стоит отметить, что реакция 3-ацетиламино-орто-карборана 3-MeC(O)NH-1,2-C2B10H11 c тетрафторборатом N-фторколлидиния [1-F-2,4,6-Me3‑NC6H2][BF4] в присутствии [(MeCN)4Pd](OTf)2 в 1,2-дихлорэтане приводит к соответствующему трифторпроизводному 3-MeC(O)NH-4,7,8-F3-1,2-C2B10H8 (схема 53 ) [105].
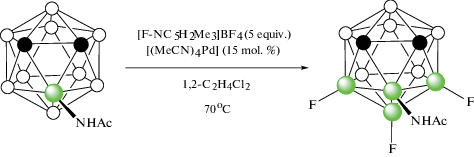
Схема 53.
Реакция 9-бензоиламино-орто-карборана 9-PhC(O)NH-1,2-C2B10H11 с арилбороновыми кислотами в присутствии Cu(OAc)2, циклогексилкарбоновой кислоты и 10 мол. % [(MeCN)4Pd][BF4] в тетрагидрофуране при 40°C приводит к соответствующим 4-арилпроизводным 9-PhC(O)NH-4-Ar-1,2-C2B10H10 (схема 54 ). Выход целевых продуктов слабо зависит от наличия в ароматическом кольце донорных или акцепторных заместителей и находится в пределах 49–74%. В качестве направляющих групп также могут использоваться другие ациламины – производные различных замещенных ароматических и алифатических кислот. К достоинствам данного метода можно отнести возможность использования незамещенного по атомам углерода карборана и широкий выбор бороновых кислот, а к недостаткам – невысокий выход как исходного 9-бензоиламино-орто-карборана, так и продуктов арилирования (особенно в случае донорных заместителей) [106].
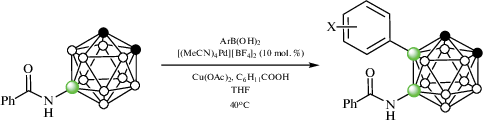
Схема 54.
Бензоиламиногруппа также может использоваться для направленного селективного окислительного сочетания двух карборановых остовов. Так, реакция 9-бензоиламино-орто-карборана 9-PhCOHN-1,2-C2B10H11 с AgF, AgOAc, 10 мол. % Pd(OAc)2 и 10 мол. % NiCl2 в толуоле при 60°С к B(4)–H-активации орто-карборана с образованием смеси диастереомерных 4,4'- и 4,5'-бис(1-(9"-бензоиламино)-орто-карборанов), которые могут быть разделены колоночной хроматографией [107].
Аналогичным образом в роли кислородсодержащей направляющей группы может выступать уреидная группа. Так, реакция N',N'-диметилуреидного производного клозо-додекабората (Bu4N)2[Me2N(O)CHNB12H11] с диарилацетиленами в присутствии Cu(OAc)2 ‧ H2O и 2.5 мол. % [Cp*RhCl2]2 в ацетонитриле при комнатной температуре приводит к B–H-активации борного остова с внедрением олефина, а также к образованию связи бор–кислород с замыканием пятичленного дибораоксазольного цикла BNCOB, приводя к (Bu4N)[1,2-NH=C(NMe2)O-3-C(R)=CH(R)-B12H9] (схема 55 ). Реакция протекает через образование комплекса [Cp*Rh(1-Me2N(O)CHNB12H9-κ3-O,H(2),H(3))], в котором атом металла координирован двумя BH-группами клозо-додекаборатного аниона (рис. 43). Реакция этого комплекса с дифенилацетиленом в присутствии Cu(OAc)2 приводит к [1,2-NH=C(NMe2)O-3-C(Ph)=CH(Ph)-B12H9]–. При этом образование 1,2-дибораоксазольного цикла “выводит из игры” направляющую группу и приводит к остановке дальнейшего замещения в клозо-додекаборатном остове. Замыкание такого цикла с образованием (Bu4N)[1,2-NH=C(NMe2)O-B12H10] также происходит при реакции (Bu4N)2[1-Me2N(O)CHNB12H11] с AgOAc в ацетонитриле [108]. Аналогичные производные с 1,2-дибораоксазольным циклом на основе клозо-додекаборатного и клозо-декаборатного анионов (Bu4N)[1,2-NH=C(R)O-B12H10] и (Bu4N)[2,1-NH=C(R)O-B10H8] образуются при обработке PhI(OAc)2 амидов (Bu4N)[R(HO)C=HN-B12H11] [109] и (Bu4N)[2-R(HO)C=HN-B10H9] [110] соответственно.
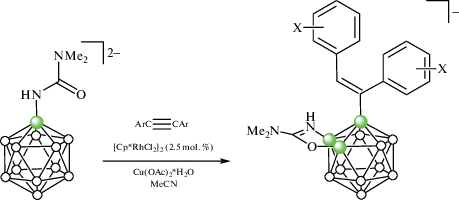
Схема 55.
Реакции B–H-активации с участием серосодержащих направляющих лигандов
Несмотря на то, что серосодержащие направляющие лиганды, такие как эфиры дитиокислот и тиоамиды, являются прямыми аналогами кислородсодержащих лигандов, целесообразно рассмотреть реакции B–H-активации с их содействием отдельно.
Реакция метилового эфира орто-карборанилдитиокарбоновой кислоты 1-MeS(S)C-1,2-C2B10H11 с [Cp*IrCl2]2 в хлористом метилене в присутствии AgOTf приводит к образованию B(3)-циклометаллированного комплекса [Cp*IrCl(1-MeS(S)C-1,2-C2B10H10-κ2-S,B(3))] с выходом 76% (схема 56 , рис. 44) [111].
Схема 56.
Рис. 44.
Строение комплекса [Cp*IrCl(1-MeS(S)C-1,2-C2B10H10-κ2-S,B(3))] (атомы водорода органических заместителей и лигандов не показаны для ясности).
При обработке S-нуклеофилами в присутствии AgOTf хлоридный лиганд в комплексе [Cp*IrCl(1-MeS(S)C-1,2-C2B10H10-κ2-S,B(3))] может быть легко заменен фенилтиолатом или орто-карборанилдитиокарбоксилатом с образованием с высоким выходом (72–85%) соответствующих комплексов [Cp*Ir(SR)(1-MeS(S)C-1,2-C2B10H10-κ2-S,B(3))] (R = Ph, 1-(S)C-1,2-C2B10H11, 1-(NPh)C-1,2-C2B10H11) (схема 56 ) [111].
Реакция [Cp*IrCl(1-MeS(S)C-1,2-C2B10H10-κ2-S,B(3))] с декабораном и триэтиламином в присутствии AgOTf в хлористом метилене приводит к образованию комплекса {(Cp*Ir(1-MeS(S)C-1,2-C2B10H10-κ2-S,B(3)))2[μ-1,10-B10H10-κ2-H,H]}, в котором роль мостикового лиганда между двумя атомами иридия выполняет клозо-декаборат-анион (рис. 45) [111].
Рис. 45.
Строение комплекса {(Cp*Ir(1-MeS(S)C-1,2-C2B10H10-κ2-S,B(3)))2[μ-1,10-B10H10-κ2-H,H]} (атомы водорода органических заместителей и лигандов не показаны для ясности).
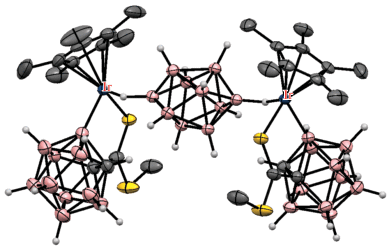
Реакция 1,12-(PhNH(S)C)2-1,12-C2B10H10 с [Cp*IrCl2]2 в присутствии AgOTf и Et3N в тетрагидрофуране приводит к B(2,6,8,9)–H-активации с образованием тетраядерного комплекса [{(Cp*Ir)2(μ-Cl)}2(1,12-S(NPh)C)2-1,12-C2B10H10-κ6-S(1),B(2);B(6);S(12),B(8);B(9))] (рис. 46) [112].
Рис. 46.
Строение комплекса [{(Cp*Ir)2(μ-Cl)}2(1,12-S(NPh)C)2-1,12-C2B10H10-κ6-S(1),B(2);B(6);S(12),B(8);B(9))] (атомы водорода органических заместителей и лигандов не показаны для ясности).
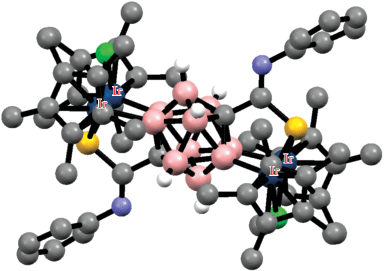
Реакция димерных комплексов [(Cp*IrCl2)2(μ-L)] (L = пиразин (pz), 4,4'-бириридин (bpy), 1,2-ди(4-пиридил)этилен (bpe), 1,4-ди(4-пиридил)бензол (dpb)) c 1,12-(PhHN(S)C)2-1,12-C2B10H10 и Et3N в хлористом метилене приводит к образованию тетраядерных металлoциклов {[(Cp*Ir)2(μ-1,12-(S(NPh)C)2-1,12-C2B10H8-κ4-S,B(2);S,B(9))](μ-L)}2, в которых B–H-активация пара-карборанового остова происходит по положениям 2 и 9 (рис. 47) [112, 113].
Рис. 47.
Строение комплексов {[(Cp*Ir)2(μ-1,12-(S(NPh)C)2-1,12-C2B10H8-κ4-S,B(2);S,B(9))](μ-pz)}2 (а) и {[(Cp*Ir)2(μ-1,12-(S(NPh)C)2-1,12-C2B10H8-κ4-S,B(2);S,B(9))](μ-dpb)}2 (б). Атомы водорода органических заместителей и лигандов не показаны для ясности.
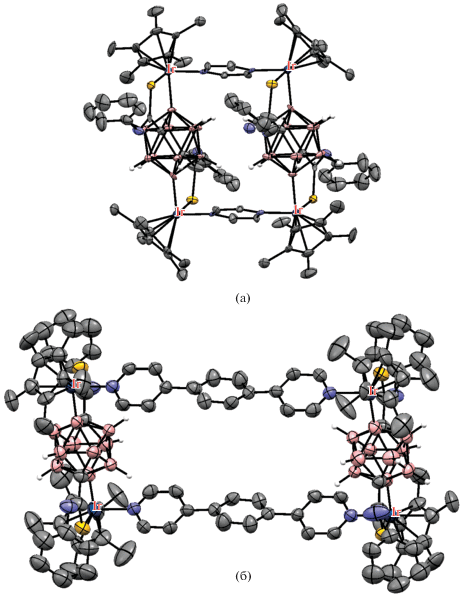
В то же время реакция димерных комплексов [(Cp*IrCl2)2(μ-L)] (L = 1,2-ди(4-пиридил)ацетилен (bpa), 2,6-ди(4-пиридил)нафталин, 2,6-ди(4-пиридил)антрацен) c 1,12-(PhHN(S)C)2-1,12-C2B10H10 и Et3N в хлористом метилене приводит к образованию тетраядерных металлoциклов {[(Cp*Ir)2(μ-1,12-(S(NPh)C)2-1,12-C2B10H8-κ4-S,B(2); S,B(8))](μ-L)}2, в которых B–H-активация пара-карборанового остова происходит по положениям 2 и 8, что подтверждается строением двухъядерного комплекса [(Cp*Ir(t-BuNC))2(μ-1,12-(S(NPh)C)2-1,12-C2B10H8-κ4-S,B(2);S,B(8))] (рис. 48), образующегося при их обработке трет-бутилизонитрилом в хлористом метилене [112].
Рис. 48.
Строение комплекса [(Cp*Ir(t-BuNC))2(μ-1,12-(PhN(S)C)2-1,12-C2B10H8-κ4-S,B(2);S,B(8))] (атомы водорода органических заместителей и лигандов не показаны для ясности).
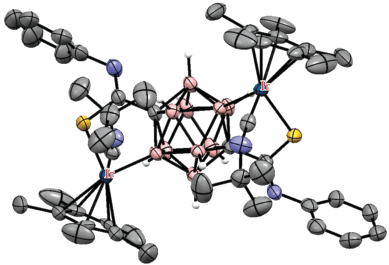
B–H-активация пара-карборанового остова по положениям 2 и 8 также наблюдается в случае комплексов {[(Cp*Ir)2(μ-1,12-(MeHN(S)C)2-1,12-C2B10H8-κ4-S,B(2);S,B(8))]2(μ-L)}(OTf)4 (L = = 1,2,3,4-тетра(4-пиридил)циклобутан) (рис. 49). Стоит отметить, что метилтиоамид, в отличие от фенилтиоамида, который может переходить в SH-имин и легко депротонироваться, не способен на это вследствие электронодонорного характера метильной группы, и благодаря этому макроциклы на его основе являются ионными [112].
ЗАКЛЮЧЕНИЕ
Анализ литературных данных демонстрирует огромный потенциал направляемой различными функциональными группами B–H-активации полиэдрических гидридов бора комплексами переходных металлов, который открывает путь к получению недоступных ранее их производных и позволяет получать комплексы необычного строения со связью бор–металл, которые могут использоваться в катализе или для создания супрамолекулярных ансамблей.
Список литературы
Sivaev I.B., Bregadze V.I. Polyhedral Boron Hydrides in Use: Current Status and Perspectives. Hauppauge: Nova Science Publishers, 2009. 85 p.
Boron Science. New Technologies and Applications / Ed. Hosmane N.S. Boca Raton: CRC Press, 2012. 878 p. https://doi.org/10.1201/b11199
Grimes R.N. Carboranes. London: Academic Press, 2016. 1058 p. https://doi.org/10.1016/B978-0-12-801894-1.09989-3
Handbook of Boron Science with Applications in Organometallics, Catalysis, Materials and Medicine / Eds. Hosmane N.S., Eagling R. London: World Scientific Publishing Europe, 2018. 1056 p. https://doi.org/10.1142/q0130
Boron-Based Compounds: Potential and Emerging Applications in Medicine / Eds. Hey-Hawkins E., Viñas Teixidor C. John Wiley & Sons Ltd., 2018. 470 p. https://doi.org/10.1002/9781119275602
Sivaev I.B., Bregadze V.I., Sjöberg S. // Collect. Czech Chem. Commun. 2002. V. 67. № 6. P. 679. https://doi.org/10.1135/cccc20020679
Sivaev I.B., Prikaznov A.V., Naoufal D. // Collect. Czech. Chem. Commun. 2010. V. 75. № 11. P. 1149. https://doi.org/10.1135/cccc2010054
Zhizhin K.Yu., Zhdanov A.P., Kuznetsov N.T. // Russ. J. Inorg. Chem. 2010. V. 55. № 14. P. 2089. https://doi.org/10.1134/S0036023610140019
Körbe S., Schreiber P.J., Michl J. // Chem. Rev. 2006. V. 106. № 12. P. 5208. https://doi.org/10.1021/cr050548u
Douvris C., Michl J. // Chem. Rev. 2013. V. 113. № 10. P. 179. https://doi.org/10.1021/cr400059k
Shmal’ko A.V., Sivaev I.B. // Russ. J. Inorg. Chem. 2019. V. 64. № 14. P. 1726. https://doi.org/10.1134/S0036023619140067
Morin J.-F., Shirai Y., Tour J.M. // Org. Lett. 2006. V. 8. № 8. P. 1713. https://doi.org/10.1021/ol060445d
Morin J.-F., Sasaki T., Shirai Y. et al. // J. Org. Chem. 2007. V. 72. № 25. P. 9481. https://doi.org/10.1021/jo701400t
Safronov A.V., Shlyakhtina N.I., Everett T.A. et al. // Inorg. Chem. 2014. V. 53. № 19. P. 10045. https://doi.org/10.1021/ic402372c
Shlyakhtina N.I., Safronov A.V., Sevryugina Yu.V. et al. // J. Organomet. Chem. 2015. V. 798. P. 234. https://doi.org/10.1016/j.jorganchem.2015.04.035
Anufriev S.A., Erokhina S.A., Suponitsky K.Yu. et al. // Eur. J. Inorg. Chem. 2017. № 38–39. P. 4444. https://doi.org/10.1002/ejic.201700575
Anufriev S.A., Timofeev S.V., Anisimov A.A. et al. // Molecules. 2020. V. 25. № 23. P. 5745. https://doi.org/10.3390/molecules25235745
Quan Y., Xie Z. // J. Am. Chem. Soc. 2014. V. 136. № 44. P. 15513. https://doi.org/10.1021/ja509557j
Quan Y., Xie Z. // Chem. Soc. Rev. 2019. V. 48. № 13. P. 3660. https://doi.org/10.1039/c9cs00169g
Hoel E.L., Hawthorne M.F. // J. Am. Chem. Soc. 1975. V. 97. № 22. P. 6388. https://doi.org/10.1021/ja00855a017
Kalinin V.N., Usatov A.V., Zakharkin L.I. // Proc. Indian Natl. Sci. Acad. A. 1989. V. 55. № 2. P. 293
Ryabov A.D. // Chem. Rev. 1990. V. 90. № 2. P. 403. https://doi.org/10.1021/cr00100a004
Albrecht M. // Chem. Rev. 2010. V. 110. № 2. P. 576. https://doi.org/10.1021/cr900279a
Lyons T.W., Sanford M.S. // Chem. Rev. 2010. V. 110. № 2. P. 1147. https://doi.org/10.1021/cr900184e
Sambiagio C., Schönbauer D., Blieck R. et al. // Chem. Soc. Rev. 2018. V. 47. № 17. P. 6603. https://doi.org/10.1039/C8CS00201K
Rej S., Das A., Chatani N. // Coord. Chem. Rev. 2021. V. 431. P. 213683. https://doi.org/10.1016/j.ccr.2020.213683
Godovikov N.N., Balema V.P., Rys E.G. // Russ. Chem. Rev. 1997. V. 66. № 12. P. 1017. [Годовиков Н.Н., Балема В.П., Рыс Е.Г. // Успехи химии. 1997. Т. 66. № 12. С. 1125.] https://doi.org/10.1070/RC1997v066n12ABEH000311
Popescu A.R., Teixidor F., Viñas C. // Coord. Chem. Rev. 2014. V. 269. P. 54. https://doi.org/10.1016/j.ccr.2014.02.016
Li K., Yao Z.-J., Deng W. // Curr. Org. Synth. 2016. V. 13. № 4. P. 504. https://doi.org/10.2174/1570179413666151218200731
Sivaev I.B., Stogniy M.Yu., Bregadze V.I. // Coord. Chem. Rev. 2021. V. 436. P. 213795. https://doi.org/10.1016/j.ccr.2021.213795
Kalinin V.N., Usatov A.V., Zakharkin L.I. // J. Organomet. Chem. 1983. V. 254. № 1. P. 127. https://doi.org/10.1016/0022-328X(83)85125-0
Ryabov A.D., Eliseev A.V., Sergeyenko E.S. et al. // Polyhedron. 1989. V. 8. № 12. P. 1485. https://doi.org/10.1016/S0277-5387(00)80324-1
Ryabov A.D., Usatov A.V., Kalinin V.N., Zakharkin L.I. // Bull. Acad. Sci. USSR, Div. Chem. Sci. 1986. V. 35. № 12. P. 2559. [Рябов А.Д., Усатов А.В., Калинин В.Н., Захаркин Л.И. // Изв. АН СССР. Сер. хим. 1986. № 12. С. 2790.] https://doi.org/10.1007/BF01474223
Fey N., Haddow M.F., Mistry R. et al. // Organometallics. 2012. V. 31. № 7. P. 2907. https://doi.org/10.1021/om201198s
Ryabov A.D., Usatov A.V., Kalinin V.N., Zakharkin L.I. // Bull. Acad. Sci. USSR, Div. Chem. Sci. 1986. V. 35. № 5. P. 1105. [Рябов А.Д., Усатов А.В., Калинин В.Н., Захаркин Л.И. // Изв. АН СССР. Сер. хим. 1986. № 5. С. 1212.] https://doi.org/10.1007/BF00955402
Zhang Z.-Y., Zhang X., Yuan J. et al. // Chem. Eur. J. 2020. V. 26. № 22. P. 5037. https://doi.org/10.1002/chem.201905647
Manojlović-Muir L., Muir K.W., Solomun T. // J. Chem. Soc., Dalton Trans. 1980. № 2. P. 317. https://doi.org/10.1039/DT9800000317
Kalinin V.N., Usatov A.V., Zakharkin L. I. // Russ. J. Gen. Chem. 1983. V. 53. № 4. P. 945. [Калинин В.Н., Усатов А.В., Захаркин Л.И. // Журн. общ. химии. 1983. Т. 53. № 4. С. 945.]
Kalinin V.N., Usatov A.V., Antonovich V.A., Zakharkin L. I. // Russ. J. Gen. Chem. 1988. V. 58. № 8. P. 1815. [Калинин В.Н., Усатов А.В., Антонович В.А., Захаркин Л.И. // Журн. общ. химии. 1988. Т. 58. № 8. С. 1815.]
Мартынова Е.В. Дис. … канд. хим. наук. М., 2007.
Usatov A.V., Martynova E.V., Dolgushin F.M. et al. // Eur. J. Inorg. Chem. 2002. № 10. P. 2565. https://doi.org/10.1002/1099-0682(200210)2002:10< 2565::AID-EJIC2565>3.0.CO;2-T
Usatov A.V., Martynova E.V., Dolgushin F.M. et al. // Eur. J. Inorg. Chem. 2003. № 1. P. 29. https://doi.org/10.1002/ejic.200390001
Estrada J., Lee S.E., McArthur S.G. et al. // J. Organomet. Chem. 2015. V. 798. № 1. P. 214. https://doi.org/10.1016/j.jorganchem.2015.05.008
Lugo C.A., Moore C.E., Rheingold A.L., Lavallo V. // Inorg. Chem. 2015. V. 54. № 5. P. 2094. https://doi.org/10.1021/ic5030636
Kalinin V.N., Usatov A.V., Zakharkin L.I. // Russ. J. Gen. Chem. 1987. V. 57. № 11. P. 2508. [Калинин В.Н., Усатов А.В., Захаркин Л.И. // Журн. общ. химии. 1987. Т. 57. № 11. С. 2508.]
Polakov A.V., Yanovskii I.A., Struchkov Yu.T. et al. // Russ. J. Сoord. Chem. 1988. V. 14. № 9. P. 1278. [Поляков А.В., Яновский А.И., Стручков Ю.Т. и др. // Коорд. химия. 1988. Т. 14. № 9. С. 1278.]
Brunner H., Apfelbacher A., Zabel M. // Eur. J. Inorg. Chem. 2001. № 4. P. 917. https://doi.org/10.1002/1099-0682(200104)2001:4< 917::AID-EJIC917>3.0.CO;2-J
Viñas C., Flores M.A., Nuñez R. et al. // Organometallics. 1998. V. 17. № 11. P. 2278. https://doi.org/10.1021/om971019m
Viñas C., Nuñez R., Teixidor F. et al. // Organometallics. 1998. V. 17. № 11. P. 2376. https://doi.org/10.1021/om970691g
Teixidor F., Nuñez R., Flores M.A. et al. // J. Organomet. Chem. 2000. V. 614-615. P. 48. https://doi.org/10.1016/S0022-328X(00)00576-3
Demonceau A., Simal F., Noels A.F. et al. // Tetrahedron Lett. 1997. V. 38. № 45. P. 7879. https://doi.org/10.1016/S0040-4039(97)10154-X
Huo X.-K., Su G., Jin G.-X. // Chem. Eur. J. 2010. V. 16. № 39. P. 12017. https://doi.org/10.1002/chem.201001278
Viñas C., Nuñez R., Flores M.A. et al. // Organometallics. 1995. V. 14. № 8. P. 3952. https://doi.org/10.1021/om00008a047
Viñas C., Nuñez R., Teixidor F. et al. // Organometallics. 1996. V. 15. № 18. P. 3850. https://doi.org/10.1021/om9509002
Demonceau A., Simal F., Noels A.F. et al. // Tetrahedron Lett. 1997. V. 38. № 23. P. 4079. https://doi.org/10.1016/S0040-4039(97)00828-9
Nuñez R., Viñas C., Teixidor F., Abad M.M. // Appl. Organomet. Chem. 2003. V. 17. № 6–7. P. 509. https://doi.org/10.1002/aoc.464
Simal F., Sebille S., Demonceau A. et al. // Tetrahedron Lett. 2000. V. 41. № 28. P. 5347. https://doi.org/10.1016/S0040-4039(00)00840-6
Eleazer B.J., Smith M.D., Peryshkov D.V. // J. Organomet. Chem. 2018. V. 829. P. 42. https://doi.org/10.1016/j.jorganchem.2016.10.040
Eleazer B.J., Smith M.D., Peryshkov D.V. // Organometallics. 2016. V. 35. № 2. P. 106. https://doi.org/10.1021/acs.organomet.5b00807
Eleazer B.J., Smith M.D., Popov A.A., Peryshkov D.V. // J. Am. Chem. Soc. 2016. V. 138. № 33. P. 10531. https://doi.org/10.1021/jacs.6b05172
Eleazer B.J., Smith M.D., Popov A.A., Peryshkov D.V. // Chem. Sci. 2018. V. 9. № 9. P. 2601. https://doi.org/10.1039/C8SC00190A
Eleazer B.J., Smith M.D., Popov A.A., Peryshkov D.V. // Chem. Sci. 2017. V. 8. № 8. P. 5399. https://doi.org/10.1039/C7SC01846K
Chan A.L., Estrada J., Kefalidis C.E., Lavallo V. // Organometallics. 2016. V. 35. № 19. P. 3257. https://doi.org/10.1021/acs.organomet.6b00622
Estrada J., Lugo C.A., McArthur S.G., Lavallo V. // Chem. Commun. 2016. V. 52. № 9. P. 1824. https://doi.org/10.1039/C5CC08377J
El-Hellani A., Kefalidis C.E., Tham F.S. et al. // Organometallics. 2013. V. 32. № 23. P. 6887. https://doi.org/10.1021/om401001p
Kalinin V.N., Usatov A.V., Zakharkin L.I. // Russ. J. Gen. Chem. 1981. V. 51. № 9. P. 2151. [Калинин В.Н., Усатов А.В., Захаркин Л.И. // Журн. общ. химии. 1981. Т. 51. № 9. С. 2151.]
Kalinin V.N., Usatov A.V., Popello I.A., Zakharkin L.I. // Bull. Acad. Sci., Chem. Sci. Div. 1982. V. 31. № 6. P. 1281. [Калинин В.Н., Усатов А.В., Попелло И.А., Захаркин Л.И. // Изв. Акад. наук СССР. Сер. хим. 1982. № 6. С. 1433.] https://doi.org/10.1007/BF00956007
Yanovskii A.I., Struchkov Yu.T., Kalinin V.N. et al. // Russ. J. Coord. Chem. 1982. V. 8. № 12. P. 1700. [Яновский А.И., Стручков Ю.Т., Калинин В.Н. и др. // Коорд. химия. 1982. Т. 8. № 12. С. 1700.]
Kalinin V.N., Usatov A.V., Zakharkin L.I. // Bull. Acad. Sci., Chem. Sci. Div. 1984. V. 33. № 7. P. 1510. [Калинин В.Н., Усатов А.В., Захаркин Л.И. // Изв. Акад. наук СССР. Сер. хим. 1984. № 7. С. 1646.] https://doi.org/10.1007/BF00956541
Zhang X., Yan H. // Chem. Sci. 2018. V. 9. № 16. P. 3964. https://doi.org/10.1039/C8SC01154K
Li C.-X., Ning Q., Zhao W. et al. // Chem. Eur. J. 2021. V. 27. № 8. P. 2699. https://doi.org/10.1002/chem.202003634
Zhang X., Zheng H., Li J. et al. // J. Am. Chem. Soc. 2017. V. 139. № 41. P. 14511. https://doi.org/10.1021/jacs.7b07160
Yanovskii A.I., Struchkov Yu.T., Kalinin V.N. et al. // Russ. J. Сoord. Chem. 1982. V. 8. № 2. P. 240. [Яновский А.И., Стручков Ю.Т., Калинин В.Н. и др. // Коорд. химия. 1982. Т. 8. № 2. С. 240.]
Gao Y., Lin Y.-J., Han Y.-F., Jin G.-X. // Dalton Trans. 2017. V. 46. № 5. P. 1585. https://doi.org/10.1039/C6DT04550B
Cui P.-F., Gao Y., Guo S.-T., Jin G.-X. // Chin. J. Chem. 2021. V. 39. № 2. P. 281. https://doi.org/10.1002/cjoc.202000461
Gao Y., Cui P.-F., Aznarez F., Jin G.-X. // Chem. Eur. J. 2018. V. 24. № 41. P. 10357. https://doi.org/10.1002/chem.201801381
Baek Y., Cheong K., Kim D., Lee P. H. // Org. Lett. 2021. V. 23. № 4. P. 1188. https://doi.org/10.1021/acs.orglett.0c04086
Yang Z., Wu Y., Fu Y. et al. // Chem. Commun. 2021. V. 57. № 13. P. 1655. https://doi.org/10.1039/d0cc07591d
Li C.-X., Zhang H.-Y., Wong T.-Y. et al. // Org. Lett. 2017. V. 19. № 19. P. 5178. https://doi.org/10.1021/acs.orglett.7b02450
Wu J., Cao K., Zhang C.-Y. et al. // Inorg. Chem. 2020. V. 59. № 23. P. 17340. https://doi.org/10.1021/acs.inorgchem.0c02638
Guo S.-T., Cui P.-F., Yuan R.-Z., Jin G.-X. // Chem. Commun. 2021. V. 57. № 19. P. 2412. https://doi.org/10.1039/D0CC08290B
Frutos M., Gomez-Gallego M., Giner E.A. et al. // Dalton Trans. 2018. V. 47. № 30. P. 9975. https://doi.org/10.1039/C8DT02296H
Chen Y., Au Y.K., Quan Y., Xie Z. // Sci. China: Chem. 2019. V. 62. № 1. P. 74. https://doi.org/10.1007/s11426-018-9388-3
Au Y.K., Lyu H., Quan Y., Xie Z. // J. Am. Chem. Soc. 2020. V. 142. № 15. P. 6940. https://doi.org/10.1021/jacs.0c02490
Chen Y., Quan Y., Xie Z. // Chem. Commun. 2020. V. 56. № 85. P. 12997. https://doi.org/10.1039/d0cc05207h
Quan Y., Tang C., Xie Z. // Chem. Sci. 2016. V. 7. № 9. P. 5838. https://doi.org/10.1039/c6sc00901h
Lyu H., Quan Y., Xie Z. // Angew. Chem. Int. Ed. 2015. V. 54. № 36. P. 10623. https://doi.org/10.1002/anie.201504481
Quan Y., Xie Z. // Angew. Chem. Int. Ed. 2016. V. 55. № 4. P. 1295. https://doi.org/10.1002/anie.201507697
Quan Y., Lyu H., Xie Z. // Chem. Commun. 2017. V. 53. № 35. P. 4818. https://doi.org/10.1039/C7CC01485F
Wang Q., Tian S., Zhang C. et al. // Org. Lett. 2019. V. 21. № 19. P. 8018. https://doi.org/10.1021/acs.orglett.9b03009
Baek Y., Cheong K., Ko G.H. et al. // J. Am. Chem. Soc. 2020. V. 142. № 22. P. 9890. https://doi.org/10.1021/jacs.0c02121
Chen Y., Quan Y., Xie Z. // Chem. Commun. 2020. V. 56. № 51. P. 7001. https://doi.org/10.1039/D0CC02531C
Au Y.K., Lyu H., Quan Y., Xie Z. // Chin. J. Chem. 2020. V. 38. № 4. P. 383. https://doi.org/10.1002/cjoc.201900475
Lin F., Yu J.-L., Shen Y. et al. // J. Am. Chem. Soc. 2018. V. 140. № 42. P. 13798. https://doi.org/10.1021/jacs.8b07872
Shen Y., Zhang K., Liang X. et al. // Chem. Sci. 2019. V. 10. № 15. P. 4177. https://doi.org/10.1039/c9sc00078j
Lyu H., Quan Y., Xie Z. // J. Am. Chem. Soc. 2016. V. 138. № 39. P. 12727. https://doi.org/10.1021/jacs.6b07086
Baek Y., Kim S., Son J.-Y. et al. // ACS Catalysis. 2019. V. 9. № 11. P. 10418. https://doi.org/10.1021/acscatal.9b03380
Han G.H., Baek Y., Lee K. et al. // Org. Lett. 2021. V. 23. № 2. P. 416. https://doi.org/10.1021/acs.orglett.0c03927
Liy H., Quan Y., Xie Z. // Angew. Chem. Int. Ed. 2016. V. 55. № 39. P. 11840. https://doi.org/10.1002/anie.201605880
Liy H., Quan Y., Xie Z. // Chem. Eur. J. 2017. V. 23. № 59. P. 14866. https://doi.org/10.1002/chem.201703006
Yao Z.-J., Yu W.-B., Lin Y.-J. et al. // J. Am. Chem. Soc. 2014. V. 136. № 7. P. 2825. https://doi.org/10.1021/ja4115665
Liang Y.-F., Yang L., Jei B.B. et al. // Chem. Sci. 2020. V. 11. № 39. P. 10764. https://doi.org/10.1039/D0SC01515F
Liang X., Shen Y., Zhang K. et al. // Chem. Commun. 2018. V. 54. № 88. P. 12451. https://doi.org/10.1039/C8CC05983G
Lian L., Lin C., Yu Y. et al. // Tetrahedron Lett. 2020. V. 61. № 51. P. 152625. https://doi.org/10.1016/tetlet.2020.152625
Lyu H., Zhang J., Yang J. et al. // J. Am. Chem. Soc. 2019. V. 141. № 10. P. 4219. https://doi.org/10.1021/jacs.9b00302
Xu T.-T., Cao K., Zhang C.-Y. et al. // Chem. Commun. 2018. V. 54. № 96. P. 13603. https://doi.org/10.1039/C8CC08193J
Wu J., Cao K., Zhang C.-Y. et al. // Org. Lett. 2019. V. 21. № 15. P. 5986. https://doi.org/10.1021/acs.orglett.9b02129
Zhang Y., Sun Y., Lin F. et al. // Angew. Chem. Int. Ed. 2016. V. 55. № 50. P. 15609. https://doi.org/10.1002/anie.201607867
Sun Y., Zhang J., Zhang Y. et al. // Chem. Eur. J. 2018. V. 24. № 41. P. 10364. https://doi.org/10.1002/chem.201801602
Voinova V.V., Selivanov N.A., Plyushchenko I.V. et al. // Molecules. 2021. V. 26. № 1. 248. https://doi.org/10.3390/molecules26010248
Yuan R.-Z., Cui P.-F., Guo S.-T., Jin G.-X. // Dalton Trans. 2021. V. 50. № 3. P. 1060. https://doi.org/10.1039/d0dt03832f
Cui P.-F., Liu X.-R., Guo S.-T. et al. // J. Am. Chem. Soc. 2021. V. 143. № 13. P. 5099. https://doi.org/10.1021/jacs.1c00779
Cui P.-F., Lin Y.-J., Li Z.-H., Jin G.-X. // J. Am. Chem. Soc. 2020. V. 142. № 18. P. 8532. https://doi.org/10.1021/jacs.0c03176
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии