

Журнал неорганической химии, 2021, T. 66, № 8, стр. 940-950
Керамика в системе K2O–CaO–SO3–P2O5
Т. В. Сафронова a, *, М. М. Ахмедов b, Т. Б. Шаталова a, С. А. Тихонова a, Г. К. Казакова a
a Московский государственный университет им. М.В. Ломоносова
119991 Москва, Ленинские горы, 1, Россия
b Российский государственный университет им. А.Н. Косыгина (Технологии. Дизайн. Искусство)
117997 Москва, Садовническая ул., 33, стр. 1, Россия
* E-mail: t3470641@yandex.ru
Поступила в редакцию 12.01.2021
После доработки 11.02.2021
Принята к публикации 15.02.2021
Аннотация
Керамика в системе K2O–CaO–SO3–P2O5 была получена из порошковых смесей, приготовленных из гидросульфата калия KНSO4 и гидроксиапатита кальция Сa10(РО4)6(ОН)2 при заданных мольных соотношениях KНSO4/Са10(РО4)6(ОН)2 = 2/1, 4/1 и 6/1. Порошковые смеси готовили в ацетоне в условиях механической активации с использованием планетарной мельницы. После гомогенизации фазовый состав порошковых смесей включал монетит CaHPO4, сингенит K2Са(SO4)2 · Н2О и гидроксиапатит кальция Сa10(РО4)6(ОН)2. После обжига в интервале температур 700–900°С фазовый состав керамики, изготовленной из порошковых смесей, включал калийзамещенный трикальцийфосфат Ca10K(PO4)7 и кальциолангбейнит K2Ca2(SO4)3, а при мольных соотношениях KНSO4/Са10(РО4)6(ОН)2 = 4/1 и 6/1 еще и сульфат калия K2SO4. Керамические материалы, фазовый состав которых включает кальциолангбейнит K2Ca2(SO4)3 и калийзамещенный трикальцийфосфат Ca10K(PO4)7, могут быть использованы в качестве резорбируемого пористого материала при лечении дефектов костной ткани методами регенеративной медицины или как матрица при создании люминесцентных/термолюминесцентных материалов. Керамические материалы в системе K2O–CaO–SO3–P2O5 были получены впервые, поэтому необходимы дополнительные исследования, определяющие оптимальное соотношение фаз для указанных областей применения.
ВВЕДЕНИЕ
Пористые неорганические матрицы, состоящие из биосовместимых биорезорбируемых фаз, представляют особый интерес для развития методов регенеративной медицины, направленных на лечение дефектов костной ткани [1]. Следует упомянуть следующие биосовместимые и биорезорбируемые фазы керамических материалов: гидроксиапатит кальция Са10(РО4)6(ОН)2, структура которого включает катионные и анионные замещения; трикальцийфосфат Са3(РО4)2; пирофосфат кальция Са2Р2О7; тромелит Ca4P6O19; полифосфат кальция Са(РО3)2; натрий- и калий-замещенные трикальцийфосфаты Ca10Na(PO4)7 и Ca10K(PO4)7; калиевый KCaPO4 и натриевый NaCaPO4 ренаниты; двойные пирофосфаты кальция/калия CaK2P2O7 и кальция/натрия CaNa2P2O7; силикат кальция CaSiO3; аморфные фазы в системах, содержащих оксиды-стеклообразователи Р2О5 и SiО2; карбонат кальция CaCO3; сульфат кальция ангидрит CaSO4.
Выбор системы K2O–CaO–SO3–P2O5 для создания резорбируемой керамики был обусловлен тем, что данной оксидной системе принадлежат такие резорбируемые и биосовместимые фазы, как калийзамещенный трикальцийфосфат Ca10K(PO4)7, калиевый ренанит KCaPO4 и сульфат кальция ангидрит CaSO4. Все эти фазы относят к биорезорбируемым и биосовместимым, поскольку при их медленном растворении выделяются биосовместимые катионы и анионы. Следует ограничивать содержание калиевого ренанита KCaPO4 в керамическом материале, предназначенном для имплантирования, поскольку гидролиз данного минерала приводит к повышению рН среды выше нейтрального значения [2]. Была получена и исследована керамика на основе сульфата кальция ангидрита CaSO4 для костных имплантатов [3–5]. Фаза сульфата кальция ангидрита СаSO4, обладающего способностью к медленному растворению [6], как правило, вводится с целью управления пределом и скоростью резорбции керамического композиционного материала, предназначенного для лечения (временной компенсации) дефекта костной ткани в процессе ее восстановления [7]. В научной литературе рассмотрено создание керамических композитов сульфат кальция ангидрит/фосфат кальция для использования в качестве костных имплантатов [8, 9].
В работах [10, 11] термическая устойчивость сульфата кальция СаSO4 указывается как возможная в интервале температур 1000–1400°С. При получении керамики, содержащей сульфат кальция СаSO4 и трикальцийфосфат Ca3(PO4)2, температура обжига, после которого фазовый состав не включал бы токсичного для организма оксида кальция СаО, указана как 1050°С [9]. Отмечается, что присутствие других солей или фаз снижает термическую устойчивость сульфата кальция ангидрита СаSO4 [6]. Таким образом, необходимо применение низкотемпературных спекающих добавок при получении керамических композиционных материалов, в состав которых планируется введение фазы сульфата кальция ангидрита СаSO4. Керамика на основе сульфата кальция ангидрита была получена с использованием спекающей добавки, представляющей собою измельченное стекло в системе SiO2–Na2O–P2O5–CaO [12].
Практически во всех статьях, посвященных получению керамики на основе сульфата кальция ангидрита СаSO4, в качестве исходного был использован порошок полуводного гипса СаSO4 · · 0.5H2O [13, 14], доступный как коммерческий реактив. Из порошка СаSO4 · 0.5H2O, обладающего вяжущими свойствами, с помощью порошковой 3D-печати формовали пористый предкерамический полуфабрикат, который затем обжигали [15].
Известны способы получения керамических материалов, фазовый состав которых формируется благодаря протеканию гетерофазных взаимодействий при термообработке [16]. При получении керамики на основе фосфатов кальция были, например, использованы порошковые смеси гидроксиапатита кальция Са10(РО4)6(ОН)2 и пирофосфата кальция Са2Р2О7, способные вступать в гетерофазное взаимодействие с образованием трикальцийфосфата Ca3(PO4)2 при нагревании (700–800°С) до начала уплотнения [17]. В настоящей работе планировали получить керамику в системе K2O–CaO–SO3–P2O5, используя в качестве исходных компонентов порошки гидроксиапатита кальция Са10(РО4)6(ОН)2 (источника оксидов кальция и фосфора) и гидросульфата калия KНSO4 (источника оксидов калия и серы), обладающего низкой температурой плавления (tпл = 210°С). Присутствие расплава, образующегося при относительно низкой температуре, позволяло предположить и возможность спекания по жидкофазному механизму, и формирование заданного фазового состава керамики в системе K2O–CaO–SO3–P2O5 вследствие гетерофазного взаимодействия компонентов порошковой системы. К сожалению, информация об этой системе в научной литературе представлена ограниченно [18], поэтому при планировании эксперимента мы опирались на известные данные о системах, относящихся к системе K2O–CaO–SO3–P2O5, таких как CaO–P2O5 [19], CaO–K2O–P2O5 [20] и K2SO4–CaSO4 [21, 22].
Целью настоящей работы было получение керамического композиционного материала в системе K2O–CaO–SO3–P2O5 на основе порошковых смесей, включающих гидроксиапатит кальция Сa10(РО4)6(ОН)2 и гидросульфат калия KНSO4 в различных мольных соотношениях.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Порошковые смеси для получения керамики готовили из гидроксиапатита кальция Ca10(PO4)6(OH)2 (CAS № 1306-06-5, puriss. p.a. ≥90%, Riedel-deHaen, Sigma-Aldrich Laborchemikalien, 04238, lot 70080, Германия) и гидросульфата калия KНSO4 (ГОСТ 4223-75, Россия). Гидросульфат калия KНSO4 (tпл = 210°С), при нагревании превращающийся в пиросульфат калия K2S2O7 (tпл = 420°С) [23], рассматривали при планировании эксперимента как компонент, способный выступать в роли добавки, обеспечивающей протекание жидкофазного спекания (включая плавление, смачивание, растекание, перегруппировку), а также способный вступать в гетерофазную реакцию, в результате которой формируется заданный фазовый состав.
Количество компонентов в исходной порошковой смеси рассчитывали по следующим реакциям, предполагая возможность их протекания при нагревании:
(1)
$\begin{gathered} {\text{С}}{{{\text{а}}}_{{10}}}{{\left( {{\text{Р}}{{{\text{О}}}_{{\text{4}}}}} \right)}_{6}}{{\left( {{\text{ОН}}} \right)}_{2}} + 2{\text{KНS}}{{{\text{O}}}_{4}} \to \\ \to {\text{2СаS}}{{{\text{O}}}_{4}} + 2{\text{С}}{{{\text{а}}}_{3}}{{\left( {{\text{Р}}{{{\text{О}}}_{4}}} \right)}_{2}} + 2{\text{KCaP}}{{{\text{O}}}_{4}} + 2{{{\text{Н}}}_{{\text{2}}}}{\text{О}}, \\ \end{gathered} $(2)
$\begin{gathered} {\text{С}}{{{\text{а}}}_{{10}}}{{\left( {{\text{Р}}{{{\text{О}}}_{4}}} \right)}_{6}}{{\left( {{\text{ОН}}} \right)}_{2}} + 4{\text{KНS}}{{{\text{O}}}_{4}} \to 3{\text{СаS}}{{{\text{O}}}_{4}} + \\ + \,\,{\text{С}}{{{\text{а}}}_{{\text{3}}}}{{\left( {{\text{Р}}{{{\text{О}}}_{{\text{4}}}}} \right)}_{2}} + 4{\text{KCaP}}{{{\text{O}}}_{4}} + {\text{S}}{{{\text{O}}}_{3}} + 3{{{\text{Н}}}_{{\text{2}}}}{\text{О}}, \\ \end{gathered} $(3)
$\begin{gathered} {\text{С}}{{{\text{а}}}_{{10}}}{{\left( {{\text{Р}}{{{\text{О}}}_{4}}} \right)}_{6}}{{\left( {{\text{ОН}}} \right)}_{2}} + 6{\text{KНS}}{{{\text{O}}}_{4}} \to \\ \to 4{\text{СаS}}{{{\text{O}}}_{4}} + 6{\text{KCaP}}{{{\text{O}}}_{4}} + 2{\text{S}}{{{\text{O}}}_{3}} + 4{{{\text{Н}}}_{{\text{2}}}}{\text{О}}. \\ \end{gathered} $Эти реакции были использованы для расчета состава исходных смесей исходя из предположения, что взаимодействие гидроксиапатита Сa10(РО4)6(ОН)2 и гидросульфата калия KНSO4 (пиросульфата калия K2S2O7) при обжиге приведет к формированию керамического материала, включающего биосовместимые фазы, такие как сульфат кальция СаSO4, трикальцийфосфат Са3(РО4)2 и калиевый ренанит KCaPO4. Допускали, что взаимодействие трикальцийфосфата Са3(РО4)2 и калиевого ренанита KCaPO4 может приводить к образованию калийзамещенного трикальцийфосфата Ca10K(PO4)7:
(4)
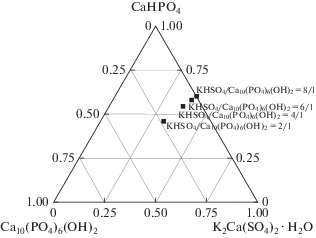
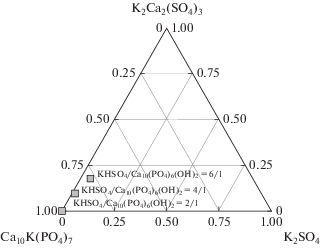
$3{\text{С}}{{{\text{а}}}_{{\text{3}}}}{{{\text{(Р}}{{{\text{О}}}_{4}})}_{2}} + {\text{KCaP}}{{{\text{O}}}_{4}} = {\text{C}}{{{\text{a}}}_{{{\text{10}}}}}{\text{K}}{{\left( {{\text{P}}{{{\text{O}}}_{{\text{4}}}}} \right)}_{7}}{\kern 1pt} .$Ожидаемый фазовый состав образцов керамики на основе порошковых смесей, состав которых был рассчитан с использованием реакций (1)–(3), а мольные соотношения KНSO4/Са10(РО4)6(ОН)2 заданы как 2/1, 4/1 и 6/1, представлен на рис. 1.
Рис. 1.
Ожидаемый фазовый состав (мол. д.) образцов керамики на основе порошковых смесей, содержащих гидроксиапатит кальция Са10(РО4)6(ОН)2 и гидросульфат калия KНSO4 при мольных соотношениях KНSO4/Са10(РО4)6(ОН)2 = 2/1, 4/1 и 6/1.

Порошки гидроксиапатита кальция Са10(РО4)6(ОН)2 и гидросульфата калия KНSO4, взятые в заданных соотношениях, мелющие тела из диоксида циркония и ацетон (ГОСТ 2603-79) были помещены в емкости из диоксида циркония. Затем емкости с порошками, мелющими телами и ацетоном закрывали и закрепляли в планетарной мельнице. Обработку порошковых смесей проводили в течение 15 мин в планетарной мельнице при соотношении порошок : мелющие тела = 1 : 5 при скорости вращения 500 об./мин.
После завершения обработки в планетарной мельнице порошковые смеси сушили на воздухе при комнатной температуре в течение 2 ч. После сушки порошковые смеси пропускали через сито с размером ячеек 200 мкм. Из полученных порошковых смесей на ручном прессе Carver Laboratory Press model (США) изготавливали компактные порошковые заготовки в форме дисков диаметром 12 мм и высотой 2–3 мм при удельном давлении прессования 100 МПа без использования временного технологического связующего. Сформованные порошковые заготовки обжигали в печи при температурах в интервале 700–900°С (скорость нагрева 5 град./мин, выдержка при заданной температуре 2 ч, охлаждение вместе с печью).
Линейную усадку и геометрическую плотность образцов керамики определяли, измерив их массу и размеры (с точностью ±0.05 мм) до и после обжига.
Рентгенофазовый анализ (РФА) порошковых смесей после обработки в условиях механической активации и образцов после обжига проводили на дифрактометре Rigaku D/Max-2500 с вращающимся анодом (Япония) с использованием CuKα-излучения. Для проведения фазового анализа использовали базу данных ICDD PDF2 [24], а также программу Match!3 (https://www.crystalimpact.com/).
Синхронный термический анализ выполняли на термоанализаторе Netzsch STA 409 PC Luxx (Netzsch, Германия) при скорости нагревания 10 град/мин. Масса образца составляла не менее 10 мг. Исследование состава образующейся при разложении образцов газовой фазы проводили при помощи квадрупольного масс-спектрометра QMS 403C Aëolos (Netzsch, Германия), совмещенного с термоанализатором Netzsch STA 409 PC Luxx. Масс-спектры записывали для массовых чисел 18 (Н2О) и 64 (SО2).
Микроструктуру образцов исследовали методом растровой электронной микроскопии на электронном микроскопе LEO SUPRA 50VP (Carl Zeiss, Германия; автоэмиссионный источник); съемку осуществляли при ускоряющем напряжении 3–20 кВ во вторичных электронах (детектор SE2). На поверхность образцов напыляли слой хрома (до 10 нм).
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
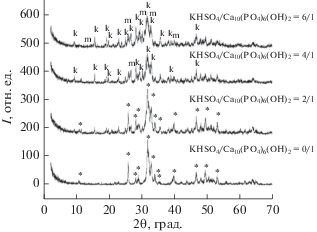
По данным РФА (рис. 2), в процессе гомогенизации в среде ацетона при использовании планетарной мельницы в порошковых смесях, в которых мольное соотношение КНSO4/Са10(РО4)6(ОН)2 составляло 2/1, 4/1 и 6/1, несмотря на использование ацетона в качестве среды дезагрегации и гомогенизации, произошло взаимодействие компонентов и изменение фазового состава. После обработки в планетарной мельнице порошковые смеси включали следующие компоненты: сингенит K2Ca(SO4)2 · · H2O, монетит CaHPO4 и гидроксиапатит кальция Са10(РО4)6(ОН)2.
Рис. 2.
Данные РФА порошковых смесей, приготовленных в условиях механической активации в ацетоне при мольных соотношениях KНSO4/Са10(РО4)6(ОН)2 = 0/1, 2/1, 4/1 и 6/1. * – Са10(РО4)6(ОН)2, PDF-Card 9-432; m – CaHPO4, PDF-Card 9-80; k – K2Ca(SO4)2 · H2O, PDF-Card 74-2423.
Формирование такого фазового состава происходило в результате взаимодействия компонентов исходной порошковой смеси, которое может быть отражено реакцией:
(5)
$\begin{gathered} {\text{С}}{{{\text{а}}}_{{{\text{10}}}}}{{\left( {{\text{Р}}{{{\text{О}}}_{{\text{4}}}}} \right)}_{{\text{6}}}}{{\left( {{\text{ОН}}} \right)}_{2}} + 8{\text{KНS}}{{{\text{O}}}_{4}} + 2{{{\text{Н}}}_{{\text{2}}}}{\text{О}} \to \\ \to 4{{{\text{K}}}_{{\text{2}}}}{\text{Са(S}}{{{\text{O}}}_{4}}{{)}_{2}}\cdot{\text{ }}{{{\text{Н}}}_{{\text{2}}}}{\text{О}} + 6{\text{CaHPO}}. \\ \end{gathered} $Сопоставление реакций (1)–(3) и (5) свидетельствует о том, что мольное соотношение KНSO4/Са10(РО4)6(ОН)2, заданное для порошковых смесей как 2/1, 4/1 и 6/1, отличается от мольного соотношения КНSO4/Са10(РО4)6(ОН)2 (8/1) для реакции (5). Гидросульфат калия KHSO4 в подготовленных смесях относительно реакции (5) взят в недостатке. Вода, необходимая для образования сингенита K2Са(SO4)2 · Н2О, частично поступала из исходных солей как в результате протекания реакции, так и будучи адсорбированной поверхностью частиц исходных порошков. Некоторое количество воды могло также поступать из ацетона, использованного в настоящем эксперименте и доступного на рынке в качестве коммерческого реагента. На рис. 3 представлены соотношения фаз, возможные при протекании реакции (5) в исследуемых смесях.
Рис. 3.
Расчетный фазовый состав (мол. д.) порошковых смесей после протекания реакции (5) между KНSO4 и Са10(РО4)6(ОН)2. Мольные соотношения исходных компонентов в порошковых смесях указаны на рисунке.
На рис. 4 представлены микрофотографии порошковых смесей после гомогенизации в ацетоне в планетарной мельнице при мольных соотношениях KНSO4/Са10(РО4)6(ОН)2, заданных как 2/1, 4/1, 6/1. На микрофотографиях можно видеть частицы с формой, близкой к изометрической, размером не более 100 нм, а также частицы в форме небольших игольчатых кристаллов длиной 100–500 нм. Кристаллы сингенита K2Са(SO4)2 · Н2О могут иметь подобную игольчатую форму [25]. Форма образовавшихся кристаллов монетита CaHPO4 близка к форме частиц исходного гидроксиапатита, на поверхности которого происходит его образование в условиях механической активации при взаимодействии солей кислой (Са(Н2РО4)2 · Н2О) и основной природы (Са10(РО4)6(ОН)2) при механической активации в ацетоне [26].
Рис. 4.
Микрофотографии порошковых смесей, приготовленных в условиях механической активации в ацетоне при мольных соотношениях KНSO4/Са10(РО4)6(ОН)2 = 2/1 (а), 4/1 (б), 6/1 (в).

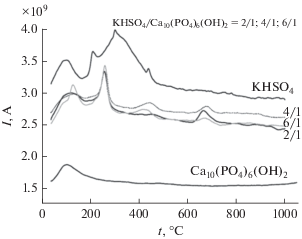
На рис. 5 представлены данные термического анализа для исходных порошков и порошковых смесей, подготовленных в условиях механической активации в ацетоне. Общая потеря массы при нагревании до 1000°С составила: для гидроксиапатита кальция – 4%; для гидросульфата калия – 32%; для порошковых смесей с мольными соотношениями компонентов KНSO4/Са10(РО4)6(ОН)2 = 2/1 – 6%; 4/1 – 12%; 6/1 – 13%. Изменение массы порошка гидроксиапатита кальция Са10(РО4)6(ОН)2 обусловлено удалением адсорбированной воды в интервале 45–180°С с пиком на кривой ионного тока для m/Z = 18 при 100°С (рис. 6). Кривая, отражающая изменение массы гидросульфата калия KНSO4, имеет несколько ступеней. До 500°С изменение массы обусловлено удалением как адсорбированной, так и химически связанной воды. А выше 500°С изменение массы может быть обусловлено выделением оксидов серы (в эксперименте ионный ток фиксировали для m/Z = 64, что соответствует выделению SO2). В интервале температур 170–500°С присутствует широкий пик сложной формы, который отражает превращение гидросульфата KHSO4 в пиросульфат калия K2S2O7 (реакция (6)) [27, 28]:
(6)
${\text{KHS}}{{{\text{O}}}_{4}} \to {{{\text{K}}}_{{\text{2}}}}{{{\text{S}}}_{{\text{2}}}}{{{\text{O}}}_{7}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}}.$Рис. 5.
Данные термического анализа исходных порошков и порошковых смесей, приготовленных в условиях механической активации в ацетоне при мольных соотношениях KНSO4/Са10(РО4)6(ОН)2 = = 2/1, 4/1, 6/1.
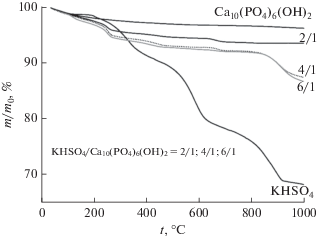
Рис. 6.
Зависимость ионного тока для m/Z = 18 (соответствует Н2О) для исходных порошков и порошковых смесей, приготовленных в условиях механической активации в ацетоне при мольных соотношениях KНSO4/Са10(РО4)6(ОН)2 = 2/1, 4/1, 6/1.
При нагревании выше 500°С, по данным [27], возможно протекание реакции (7):
(7)
${{{\text{S}}}_{2}}{\text{O}}_{7}^{{2 + }} \to {\text{S}}{{{\text{O}}}_{3}} + {\text{SO}}_{4}^{{2 - }}.$Выделение SO2, который рассматривается как дублер SO3, для гидросульфата калия KHSO4 зафиксировано на кривой ионного тока для m/Z = = 64 (SO2) выше 400°С (рис. 7).
Рис. 7.
Зависимость ионного тока для m/Z = 64 (соответствует SO2) для исходных порошков и порошковых смесей, приготовленных в условиях механической активации в ацетоне при мольных соотношениях KНSO4/Са10(РО4)6(ОН)2 = 2/1, 4/1, 6/1.
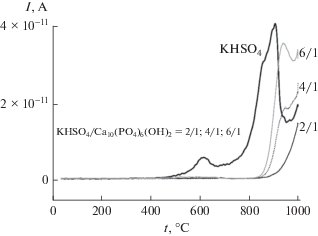
Зависимости массы порошковых смесей от температуры отличаются от кривых для исходных компонентов. Изменение массы порошковых смесей с различным мольным соотношением исходных компонентов в значительной степени обусловлено потерей адсорбированной воды, а также разложением образовавшихся при обработке в планетарной мельнице минералов, а именно сингенита K2Са(SO4)2 · Н2О (~250°С, реакция (8)) [29] и монетита CaHPO4 (~400°С, реакция (9)) [26]. Согласно [29], синтетический сингенит подвергается дегидратации и разложению (реакция (10)) при нагревании выше 240°С:
(8)
${{{\text{K}}}_{{\text{2}}}}{\text{Са(S}}{{{\text{O}}}_{{\text{4}}}}{{)}_{2}}\cdot{\text{ }}{{{\text{Н}}}_{{\text{2}}}}{\text{О}} \to {{{\text{K}}}_{{\text{2}}}}{\text{Са(S}}{{{\text{O}}}_{{\text{4}}}}{{)}_{2}} + {{{\text{Н}}}_{{\text{2}}}}{\text{О}},$(9)
$2{\text{CaHP}}{{{\text{O}}}_{4}} \to {\text{C}}{{{\text{a}}}_{{\text{2}}}}{{{\text{P}}}_{{\text{2}}}}{{{\text{O}}}_{7}} + {{{\text{Н}}}_{{\text{2}}}}{\text{О}},$(10)
$2{{{\text{K}}}_{{\text{2}}}}{\text{Са(S}}{{{\text{O}}}_{{\text{4}}}}{{)}_{2}} \to {{{\text{K}}}_{{\text{2}}}}{\text{C}}{{{\text{a}}}_{{\text{2}}}}{{{\text{(S}}{{{\text{O}}}_{4}})}_{3}} + {{{\text{K}}}_{{\text{2}}}}{\text{S}}{{{\text{O}}}_{4}}.$Таким образом, можно предположить, что в порошковых смесях после 400°С присутствуют следующие компоненты: не вступивший в реакцию при механической активации гидроксиапатит кальция Са10(РО4)6(ОН)2; пирофосфат кальция Ca2P2O7, образовавшийся в результате термической конверсии монетита CaHPO4; кальциолангбейнит K2Ca2(SO4)3 и сульфат калия K2SO4, образовавшиеся из сингенита K2Са(SO4)2 · Н2О. При повышении температуры данные компоненты взаимодействуют друг с другом и формируют фазовый состав керамических материалов.
Изменение массы в интервалах 620–750°С для порошковой смеси с мольным соотношением KНSO4/Са10(РО4)6(ОН)2 = 2/1, 630–740°С для порошковой смеси с мольным соотношением KНSO4/Са10(РО4)6(ОН)2 = 4/1 и 635–730°С для порошковой смеси с мольным соотношением КНSO4/Са10(РО4)6(ОН)2 = 6/1 обусловлено выделением воды (рис. 6), которая, по всей видимости, формируется в результате протекания реакции (11). Ступень на кривой изменения массы для порошковой смеси с мольным соотношением КНSO4/Са10(РО4)6(ОН)2 = 6/1 практически незаметна (рис. 5), а интервал выделения воды наименьший (рис. 6), поскольку в этом порошке после обработки в условиях механической активации количество не вступившего в реакцию гидроксиапатита кальция минимальное (рис. 3):
(11)
$\begin{gathered} {\text{С}}{{{\text{а}}}_{{10}}}{{\left( {{\text{Р}}{{{\text{О}}}_{4}}} \right)}_{6}}{{\left( {{\text{ОН}}} \right)}_{2}} + {\text{C}}{{{\text{a}}}_{{\text{2}}}}{{{\text{P}}}_{{\text{2}}}}{{{\text{O}}}_{7}} \to \\ \to 4{\text{C}}{{{\text{a}}}_{3}}{{({\text{P}}{{{\text{O}}}_{4}})}_{2}} + {{{\text{Н}}}_{{\text{2}}}}{\text{О}}. \\ \end{gathered} $Заметная потеря массы порошковых смесей с мольными соотношениями KHSO4/Са10(РО4)6(ОН)2 = = 4/1 и 6/1 наблюдается выше 830°С (рис. 5). Уменьшение массы порошковых смесей с мольными соотношениями KНSO4/Са10(РО4)6(ОН)2 = = 4/1 и 6/1 на 5 и 6% соответственно обусловлено выделением оксидов серы (зафиксировано для SO2, m/Z = 64, рис. 7). Для порошковой смеси KНSO4/Са10(РО4)6(ОН)2 = 2/1 температура начала выделения оксида серы составляет 865°С, для порошковой смеси KНSO4/Са10(РО4)6(ОН)2 = 4/1 – 850°С, а для смеси КНSO4/Са10(РО4)6(ОН)2 = = 6/1 – 830°С (рис. 7). Выделение SO2, по всей видимости, связано с возможным испарением и разложением сульфата калия из расплава, поскольку значения данных температур близки к температуре эвтектики (867°С) в системе CaSO4–K2SO4 [21].
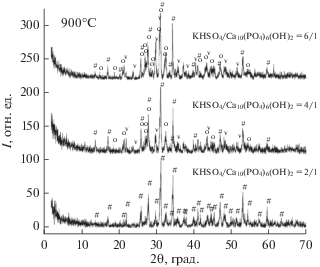
Фазовый состав керамики (рис. 8) на основе порошковой смеси КНSO4/Са10(РО4)6(ОН)2 с мольным соотношением 2/1 после обжига в интервале температур 700–900°C был представлен калийзамещенным трикальцийфосфатом Ca10K(PO4)7, на основе порошковых смесей с мольным соотношением КНSO4/Са10(РО4)6(ОН)2 = 4/1 и 6/1 – калий-замещенным трикальцийфосфатом Ca10K(PO4)7, кальциолангбейнитом (двойным сульфатом калия и кальция) K2Ca2(SO4)3 и сульфатом калия K2SO4.
Рис. 8.
Данные РФА керамики из порошковых смесей, приготовленных в условиях механической активации в ацетоне при мольных соотношениях KНSO4/ Са10(РО4)6(ОН)2 = 2/1, 4/1, 6/1, после обжига при 900°С: # – Ca10K(PO4)7 (карточка PDF 45-138); o – K2Ca2(SO4)3 (карточка PDF 20-867); v – K2SO4, (карточка PDF 5-613).
Образование фазы кальциолангбейнита K2Ca2(SO4)3 и сульфата калия K2SO4 из K2Са(SO4)2 при нагревании может быть отражено реакцией (10), фаз калийзамещенного трикальцийфосфата Ca10K(PO4)7 и кальциолангбейнита K2Ca2(SO4)3 – реакциями (12) и (13) или реакцией (14):
(12)
$\begin{gathered} 7{\text{C}}{{{\text{a}}}_{{\text{3}}}}{{\left( {{\text{P}}{{{\text{O}}}_{{\text{4}}}}} \right)}_{2}} + {{{\text{K}}}_{{\text{2}}}}{\text{S}}{{{\text{O}}}_{4}} \to \\ \to 2{\text{C}}{{{\text{a}}}_{{{\text{10}}}}}{\text{K}}{{\left( {{\text{P}}{{{\text{O}}}_{{\text{4}}}}} \right)}_{7}} + {\text{CaS}}{{{\text{O}}}_{4}}, \\ \end{gathered} $(13)
${{{\text{K}}}_{{\text{2}}}}{\text{Са(S}}{{{\text{O}}}_{{\text{4}}}}{{{\text{)}}}_{2}} + {\text{CaS}}{{{\text{O}}}_{4}} \to {{{\text{K}}}_{{\text{2}}}}{\text{C}}{{{\text{a}}}_{{\text{2}}}}{{{\text{(S}}{{{\text{O}}}_{4}})}_{3}},$(14)
$\begin{gathered} 7{\text{C}}{{{\text{a}}}_{3}}{{\left( {{\text{P}}{{{\text{O}}}_{4}}} \right)}_{2}} + 3{{{\text{K}}}_{{\text{2}}}}{\text{Са(S}}{{{\text{O}}}_{4}}{{)}_{2}} \to \\ \to 2{\text{C}}{{{\text{a}}}_{{{\text{10}}}}}{\text{K}}{{\left( {{\text{P}}{{{\text{O}}}_{{\text{4}}}}} \right)}_{7}} + 2{{{\text{K}}}_{{\text{2}}}}{\text{C}}{{{\text{a}}}_{{\text{2}}}}{{{\text{(S}}{{{\text{O}}}_{4}})}_{3}}. \\ \end{gathered} $На рис. 9 представлено количественное соотношение фаз (мас. %) в керамике из порошковых смесей, подготовленных в условиях механической активации в ацетоне при мольных соотношениях КНSO4/Са10(РО4)6(ОН)2 = 2/1, 4/1, 6/1 после обжига при 900°С. Количественное соотношение фаз было определено с помощью программы Match!3, https://www.crystalimpact.com/. Этот рисунок наглядно показывает, что фазовый состав полученных керамических материалов представлен преимущественно двумя фазами – калийзамещенным трикальцийфосфатом Ca10K(PO4)7 и кальциолангбейнитом K2Са2(SO4)3. Биосовместимость и резорбируемость калийзамещенного трикальцийфосфата Ca10K(PO4)7 известна из литературы [30]. Кальциолангбейнит K2Са2(SO4)3 применяли ранее как матрицу для создания люминесцентных/термолюминесцентных материалов при допировании европием Eu или медью Cu [31, 32]. Образование кальциолангбейнита K2Са2(SO4)3 возможно при производстве цементного клинкера [22]. Стекла в системе K2O–CaO–SO3–P2O5 рассматривали как возможный компонент минеральных удобрений [18]. Кальциолангбейнит K2Са2(SO4)3 входит в состав цемента MTA-Angelus® (https://www.angelusdental.com/), предназначенного для лечения зубов [33], что, как можно предположить, указывает на биосовместимость этого минерала. Тем не менее необходимо провести дополнительные исследования биосовместимости in vitro и in vivo керамических материалов, содержащих в значительном количестве фазу кальциолангбейнита K2Са2(SO4)3.
Рис. 9.
Фазовый состав керамики (мас. %) из порошковых смесей, приготовленных в условиях механической активации в ацетоне при мольных соотношениях KНSO4/Са10(РО4)6(ОН)2 = 2/1, 4/1, 6/1, после обжига при 900°С. Использованы данные о количественном соотношении фаз, полученные с использованием программы Match!3 (https://www.crystalimpact.com/).
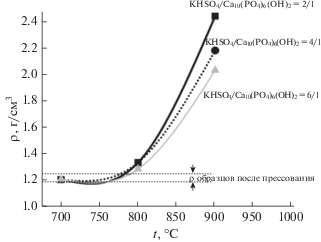
На рис. 10 представлена зависимость от температуры диаметра образцов порошковых смесей, приготовленных в условиях механической активации в ацетоне при мольных соотношениях КНSO4/Са10(РО4)6(ОН)2 = 2/1, 4/1, 6/1. Для образца порошковой смеси с мольным соотношением КНSO4/Са10(РО4)6(ОН)2 = 2/1 усадка составила 27%, для образца с КНSO4/Са10(РО4)6(ОН)2 = = 4/1 усадка после обжига при 900°С составила 23%, для образца с мольным соотношением КНSO4/Са10(РО4)6(ОН)2 = 6/1 усадка составила 21%. Данные о фазовом составе полученных образцов указывают (из-за присутствия K2SO4 или эвтектики (867°С) в системе CaSO4–K2SO4 [21]) на возможность спекания по жидкофазному механизму. При этом выделение оксидов серы при обжиге выше температуры эвтектики (867°С) в системе CaSO4–K2SO4 для порошковых смесей с мольным соотношением КНSO4/Са10(РО4)6(ОН)2 = = 4/1 и 6/1 несколько препятствует уплотнению (рис. 7). Достигнутая плотность 2.4, 2.1 и 2.0 г/см3 (рис. 11) для образцов керамики из порошков, приготовленных при мольном соотношении КНSO4/Са10(РО4)6(ОН)2 = 2/1, 4/1 и 6/1 соответственно, после обжига при 900°С значительно меньше, чем расчетная плотность для кальциолангбейнита K2Са2(SO4)3 (2.74 г/см3 [34]) или для калийзамещенного трикальцийфосфата Ca10K(PO4)7 (3.11 г/см3 [35]). Внешний вид образца керамики из порошка, приготовленного при мольном соотношении KНSO4/Са10(РО4)6(ОН)2 = = 6/1 (рис. 10), имеет шероховатую поверхность, которая вполне могла сформироваться при выделении газообразного продукта.
Рис. 10.
Зависимость от температуры обжига диаметра образцов керамики из порошковых смесей, приготовленных в условиях механической активации в ацетоне при мольных соотношениях KНSO4/ Са10(РО4)6(ОН)2 = 2/1, 4/1, 6/1.
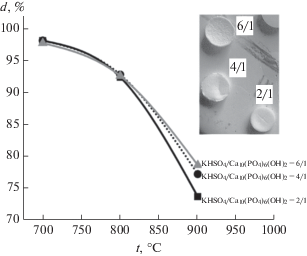
Рис. 11.
Зависимость от температуры обжига плотности образцов керамики из порошковых смесей, приготовленных в условиях механической активации в ацетоне при мольных соотношениях KНSO4/ Са10(РО4)6(ОН)2 = 2/1, 4/1, 6/1.
На рис. 12 представлены микрофотографии сколов образцов керамики из порошковых смесей, подготовленных в условиях механической активации в ацетоне при мольных соотношениях KНSO4/Са10(РО4)6(ОН)2 = 2/1 (а, б), 4/1 (в, г), 6/1 (д, е), после обжига при 900°С. Микроструктура образцов керамики в системе K2O–CaO–SO3–P2O5 существенным образом зависит от состава использованных порошковых смесей. При относительно малом увеличении видно, что образцы керамики из порошковой смеси, приготовленной в условиях механической активации в ацетоне при мольном соотношении KНSO4/Са10(РО4)6(ОН)2 = = 2/1, обладают пористостью с размером пор 3–10 мкм (рис. 12а). Образцы керамики из порошковой смеси, подготовленной в условиях механической активации в ацетоне при мольном соотношении KНSO4/Са10(РО4)6(ОН)2 = 4/1, обладают пористостью с размером пор 20–60 мкм (рис. 12в). А образцы керамики из порошковых смесей, подготовленных в условиях механической активации в ацетоне при мольном соотношении KНSO4/Са10(РО4)6(ОН)2 = 6/1 вообще выглядят не спеченными (рис. 12д, 12е).
Рис. 12.
Микрофотографии сколов образцов керамики из порошковых смесей, приготовленных в условиях механической активации в ацетоне при мольных соотношениях KНSO4/Са10(РО4)6(ОН)2 = 2/1 (а, б), 4/1 (в, г), 6/1 (д, е), после обжига при 900°С.

Размеры зерен в керамике на основе порошковой смеси с мольным соотношением КНSO4/Са10(РО4)6(ОН)2 = 2/1 составляют 0.2–0.5 мкм, на основе порошковой смеси с мольным соотношением КНSO4/Са10(РО4)6(ОН)2 = 4/1 – 0.5–1 мкм, а с мольным соотношением 6/1 – от 0.5–2 до 10 мкм.
ЗАКЛЮЧЕНИЕ
Для получения керамики в системе K2O–CaO–SO3–P2O5 были использованы обработанные в ацетоне в условиях механической активации в планетарной мельнице порошковые смеси, приготовленные из гидроксиапатита кальция Сa10(РО4)6(ОН)2 (источника оксидов кальция и фосфора) и гидросульфата калия KНSO4 (источника оксидов калия и серы) при различных мольных соотношениях KНSO4/Са10(РО4)6(ОН)2, заданных как 2/1, 4/1 и 6/1. При проведении настоящего исследования впервые была установлена возможность протекания химической реакции в условиях механической активации в ацетоне между гидроксиапатитом кальция Сa10(РО4)6(ОН)2 и гидросульфатом калия KНSO4 с образованием сингенита K2Са(SO4)2 · Н2О и монетита CaHPO4. Данные термического и рентгенофазового анализа позволяют утверждать, что формирование фазового состава керамических материалов в системе K2O–CaO–SO3–P2O5 из подготовленных порошковых смесей происходит при нагревании в результате взаимодействия гидроксиапатита кальция Сa10(РО4)6(ОН)2, пирофосфата кальция Са2Р2О7, кальциолангбейнита K2Ca2(SO4)3 и сульфата калия K2SO4. Получены образцы керамики, фазовый состав которых представлен в основном калийзамещенным трикальцийфосфатом Ca10K(PO4)7 и кальциолангбейнитом K2Ca2(SO4)3. Образующийся в результате термического разложения сингенита K2Са(SO4)2 · Н2О сульфат калия K2SO4 (tпл = 1069°С, tэвт = 867°С в системе K2SO4–Са2SO4) может быть рассмотрен как спекающая добавка, позволяющая получать керамику при относительно невысокой (900°С) температуре обжига. Керамические материалы, фазовый состав которых включает кальциолангбейнит K2Ca2(SO4)3 и калийзамещенный трикальцийфосфат Ca10K(PO4)7, могут быть использованы в качестве резорбируемой пористой матрицы при лечении дефектов костной ткани методами регенеративной медицины или как основа люминесцентных/термолюминесцентных материалов. Керамические материалы в системе K2O–CaO–SO3–P2O5 были получены впервые, поэтому необходимы дополнительные исследования, определяющие оптимальное соотношение фаз для указанных областей применения.
Список литературы
Pina S., Ribeiro V.P., Marques C.F. et al. // Materials. 2019. V. 12. № 11. P. 1824. https://doi.org/10.3390/ma12111824
Orlov N.K., Putlayev V.I., Evdokimov P.V. et al. // Inorg. Mater. 2018. V. 54. № 5. P. 500. [Орлов Н.К., Путляев В.И., Евдокимов П.В. и др. // Неорган. материалы. 2018. Т. 54. № 5. С. 523.]https://doi.org/10.1134/S0020168518050096
Chang M.P., Hsu H.C., Tuan W.H. et al. // J. Med. Biol. Eng. 2017. V. 37. № 6. P. 879. https://doi.org/10.1007/s40846-017-0253-1
Zhou J., Gao C., Feng P. et al. // J. Porous Mater. 2015. V. 22. № 5. P. 1171. https://doi.org/10.1007/s10934-015-9993-x
Chang H.Y., Chen Y.C., Hsu P.Y. et al. // Adv. Powder. Technol. 2020. V. 31. № 10. P. 4180. https://doi.org/10.1016/j.apt.2020.08.023
Freyer D., Voigt W. // Monatsh. Chem. 2003. V. 134. № 5. P. 693. https://doi.org/10.1007/s00706-003-0590-3
Zhou J., Yuan F., Peng S. et al. // Appl. Sci. 2016. V. 6. № 12. P. 411. https://doi.org/10.3390/app6120411
Yang D., Yang Z., Li X. et al. // Ceram. Int. 2005. V. 31. № 7. P. 1021. https://doi.org/10.1016/j.ceramint.2004.10.016
Yang Z., Yang D.A., Zhao H. // Key Eng. Mater. 2007. V. 336. P. 1635. https://doi.org/10.4028/www.scientific.net/kem.336-338.1635
Ostroff A.G., Sanderson R.T. // J. Inorg. Nucl. Chem. 1959. V. 9. № 1. P. 45. https://doi.org/10.1016/0022-1902(59)80009-9
Collier N.C. // Ceramics-Silikaty. 2016. V. 60. № 4. P. 338. https://doi.org/10.13168/cs.2016.0050
Chang M.P., Tsung Y.C., Hsu H.C. et al. // Ceram. Int. 2015. V. 41. № 1. P. 1155. https://doi.org/10.1016/j.ceramint.2014.09.043
Kuo S.T., Wu H.W., Tuan W.H. et al. // J. Mater. Sci: Mater. Med. 2012. V. 23. № 10. P. 2437. https://doi.org/10.1007/s10856-012-4704-5
Hsu P.Y., Chang M.P., Tuan W.H. et al. // Ceram. Int. 2018. V. 44. № 8. P. 8934. https://doi.org/10.1016/j.ceramint.2018.02.088
Dikici B.A., Dikici S., Karaman O. et al. // Biocybern. Biomed. Eng. 2017. V. 37. № 4. P. 733. https://doi.org/10.1016/j.bbe.2017.08.007
Iqbal Y., Lee W.E. // J. Am. Ceram. Soc. 1999. V. 82. № 12. P. 3584. https://doi.org/10.1111/j.1151-2916.1999.tb02282.x
Safronova T., Putlayev V., Shekhirev M. // Powder Metall. Met. Ceram. 2013. V. 52. № 5-6. P. 357. https://doi.org/10.1007/s11106-013-9534-6
Ghosh K., DasMohapatra G.K., Soodbiswas N. // Phys. Chem. Glasses. 2003. V. 44. № 4. P. 313. https://www.ingentaconnect.com/content/sgt/pcg/2003/ 00000044/00000004/art00010
Ding G.H., Xie W., Jung I.H. et al. // Acta Phys.-Chim. Sin. 2015. V. 31. № 10. C. 1853. https://doi.org/10.3866/PKU.WHXB201508121
Sandström M.H., Boström D. // J. Chem. Thermodyn. 2008. V. 40. № 1. P. 40. https://doi.org/10.1016/j.jct.2007.06.006
Rowe J.J., Morey G.W., Hansen I.D. // J. Inorg. Nucl. Chem. 1965. V. 27. № 1. P. 53. https://doi.org/10.1016/0022-1902(65)80189-0
Arceo H.B., Glasser F.P. // Cem. Concr. Res. 1990. V. 20. № 6. P. 862. https://doi.org/10.1016/0008-8846(90)90047-2
Eriksen K.M., Fehrmann R., Hatem G. et al. // J. Phys. Chem. 1996. V. 100. № 25. P. 10771. https://doi.org/10.1021/jp953744l
ICDD (2010). PDF-4+ 2010 (Database), edited by Dr. Soorya Kabekkodu, International Centre for Diffraction Data. Newtown Square. PA. USA. http://www.icdd.com/products/pdf2.htm
Matović V., Erić S., Kremenović A. et al. // J. Cult. Herit. 2012. V. 13. № 2. P. 175. https://doi.org/10.1016/j.culher.2011.09.003
Safronova T.V., Sadilov I.S., Chaikun K.V. et al. // Russ. J. Inorg. Chem. 2019. V. 64. № 9. P. 1088. [Сафронова Т.В., Садилов И.С., Чайкун К.В. и др. // Журн. неорган. химии. 2019. Т. 64. № 9. С. 916.]https://doi.org/10.1134/S0036023619090171
Fehrmann R., Hansen N.H., Bjerrum N.J. // Inorg. Chem. 1983. V. 22. № 26. P. 4009. https://doi.org/10.1021/ic00168a038
Diosa J.E., Vargas R.A., Mina E. et al. // Phys. Status Solidi B. 2000. V. 220. № 1. P. 641. https://doi.org/10.1002/1521-3951(200007)220:1<641::AID-PSSB641>3.0.CO;2-X
Kloprogge J.T., Ding Z., Martens W.N. et al. // Thermochim. Acta. 2004. V. 417. № 1. P. 143. https://doi.org/10.1016/j.tca.2003.12.001
Radetzki F., Wohlrab D., Zeh A. et al. // Biomed. Mater. Eng. 2011. V. 21. № 5-6. P. 307. https://doi.org/10.3233/BME-2012-0678
Pandey A., Sonkawade R.G., Sahare P.D. // J. Phys. D: Appl. Phys. 2002. V. 35. № 21. P. 2744. https://doi.org/10.1088/0022-3727/35/21/309
Sahare P.D., Bakare J.S., Dhole S.D. et al. // Radiat. Meas. 2012. V. 47. № 11-12. P. 1083. https://doi.org/10.1016/j.radmeas.2012.10.003
Garcia A., de Lima Machado M.E., Britto M.L.B. et al. // J. Health. Sci. Inst. 2011. V. 29. № 2. P. 89. https://www.unip.br/presencial/comunicacao/publicacoes/ics/edicoes/2011/02_abr-jun/V29_n2_2011_p89-91.pdf
Pekov I.V., Zelenski M.E., Zubkova N.V. et al. // Mineral. Mag. 2012. V. 76. № 3. P. 673. https://doi.org/10.1180/minmag.2012.076.3.16
Oralkov S.Yu., Lazoryak B.I., Aziev R.G. // Russ. J. Inorg. Chem. 1988. V. 33. № 1. С. 73. [Оралков С.Ю., Лазоряк Б.И., Азиев Р.Г. // Журн. неорган. химии. 1988. Т. 33. № 1. С. 73.]
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии