

Журнал неорганической химии, 2021, T. 66, № 5, стр. 610-615
Синтез оксида Ba0.5Sr0.5Co0.8Fe0.2O3 – δ, перспективного в качестве катодного материала современных твердооксидных топливных элементов
Т. Л. Симоненко a, *, Н. П. Симоненко a, Е. П. Симоненко a, В. Г. Севастьянов a, Н. Т. Кузнецов a
a Институт общей и неорганической химии им. Н.С. Курнакова РАН
119991 Москва, Ленинский пр-т, 31, Россия
* E-mail: egorova.offver@gmail.com
Поступила в редакцию 01.12.2020
После доработки 28.12.2020
Принята к публикации 29.12.2020
Аннотация
Изучен процесс синтеза твердого раствора состава Ba0.5Sr0.5Co0.8Fe0.2O3–δ с использованием гликоль-цитратного метода. В рамках синхронного термического анализа в потоке воздуха в интервале температур 25–1200°С исследовано термическое поведение полученного порошка и определены условия термообработки с целью получения оксида с заданной кубической кристаллической структурой типа перовскита. С помощью комплекса современных методов физико-химического анализа (рентгенофазового и рентгеноспектрального элементного анализа, ИК-спектроскопии) проведена аттестация полученного порошка, исследован его фазовый состав и микроструктура. Показано, что предложенный метод и условия синтеза эффективны для получения однофазного оксида указанного состава, не содержащего примесных включений.
ВВЕДЕНИЕ
На сегодняшний день в связи с возрастающей потребностью в современных экологически чистых источниках электроэнергии ведутся активные разработки с целью получения новых материалов и технологий для создания эффективных устройств альтернативной энергетики [1, 2], в частности электрохимических генераторов энергии [3]. Среди них следует отметить твердооксидные топливные элементы (ТОТЭ), характеризующиеся наиболее высоким КПД и способные использовать в качестве топлива как водород разной чистоты, так и различные углеводороды (метан, природный газ, биотопливо и др.) [4–7]. Одной из ключевых задач при разработке ТОТЭ является снижение их рабочих температур и создание среднетемпературных топливных ячеек, не уступающих по своим рабочим характеристикам классическим высокотемпературным устройствам на основе твердых электролитов в системе ZrO2–Y2O3. Такой переход позволит существенно расширить круг возможных конструкционных материалов, увеличить срок службы и стоимость всего топливного элемента в целом. Достижение указанной цели на сегодняшний день возможно при реализации двух подходов. Это разработка и получение среднетемпературных материалов с различным типом проводимости (в том числе использование протонных проводников) в качестве компонентов ТОТЭ, а также снижение толщины функциональных слоев и формирование топливной ячейки планарной геометрии [8–11]. Одной из важнейших проблем при снижении рабочих температур ТОТЭ является повышение поляризационного сопротивления на катоде, возникающее в связи со сниженной концентрацией кислородных вакансий при температурах ниже 800°С, в результате чего наблюдается также ухудшение каталитической активности данного электрода [12–14]. Особое внимание привлекают кобальт- и железосодержащие оксиды, в частности BaxSr1 –xCoyFe1 –yO3 – δ, со структурой типа перовскита, обладающие наибольшей величиной (по сравнению с другими типами катодных материалов) электронно-ионной проводимости, электрокаталитической активностью, а также довольно низким (0.51–0.60 Ом см2 [15]) поляризационным сопротивлением [16–18]. Кроме того, катоды подобного состава могут использоваться в топливных ячейках как с кислород-ионным [19, 20], так и с протонным твердым электролитом [21]. В литературе имеется ряд публикаций по получению порошков состава BaxSr1 –xCo0.8Fe0.2O3 – δ (0.3 ≤ x ≤ 0.7) с использованием различных методов: твердофазного синтеза [22–24], золь-гель технологии [25, 26], совместного осаждения [27, 28], спрей-пиролиза [29]. При этом необходимо отметить, что использование вышеперечисленных методов с целью получения многокомпонентных твердых растворов и соединений сопряжено со значительным риском разделения фаз и отклонением состава от целевого. Среди наиболее удобных и масштабируемых методов синтеза оксидов сложного состава, позволяющих контролировать дисперсность и микроструктуру получаемых продуктов благодаря варьированию соотношения между неорганическими солями и органическими компонентами, следует выделить методы сжигания: цитрат-нитратный, глицин-нитратный и гликоль-цитратный [30–34].
Таким образом, цель настоящей работы – изучение процесса гликоль-цитратного синтеза оксида состава Ba0.5Sr0.5Co0.8Fe0.2O3 – δ, а также исследование его термического поведения, фазового состава и микроструктурных особенностей.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Нанопорошок состава Ba0.5Sr0.5Co0.8Fe0.2O3 – δ был получен гликоль-цитратным методом (рис. 1). В качестве исходных реагентов использовали нитраты бария, кобальта, стронция и хлорид железа, навески которых были растворены в минимальном количестве дистиллированной воды. С целью предотвращения загрязнения целевого оксида ионами хлора непосредственно перед началом синтеза проводили кипячение растворов солей в присутствии азотной кислоты, в результате чего хлорид-ионы замещались на N${\text{O}}_{3}^{ - }$-группы. Далее к полученной реакционной смеси добавляли моногидрат лимонной кислоты, нитрат аммония и этиленгликоль в мольном соотношении к целевому продукту 3 : 1, 6 : 1 и 2 : 1 соответственно. Полученную смесь упаривали при 250°С до образования вязкой системы, которая при дальнейшем нагреве самовоспламенялась с образованием целевого вспененного оксидного нанопорошка.

Термическое поведение полученного нанопорошка было изучено с помощью синхронного (ТГА/ДСК) термического анализа на термоанализаторе SDT Q-600. Контролируемый нагрев проводили в Al2O3-микротиглях в интервале температур 20–1200°С со скоростью 20 град/мин в потоке воздуха 250 мл/мин.
ИК-спектр пропускания исследуемого порошка (после нагрева до 1200°С) был записан с использованием ИК-Фурье-спектрометра ИнфраЛЮМ ФТ-08. Была приготовлена суспензия порошка в вазелиновом масле, которую помещали в виде пленки между стеклами KBr. Спектральный анализ проводили в диапазоне волновых чисел 350–4000 см–1 (время накопления сигнала составляло 15 с, разрешение – 1 см–1).
Рентгенофазовый анализ (РФА) порошка выполняли на дифрактометре Bruker D8 Advance (CuKα-излучение, диапазон углов 2θ 20°–80°, разрешение 0.02°, время накопления сигнала в точке 0.3 с).
Микроструктуру и элементный состав полученного порошка изучали методом растровой электронной микроскопии (РЭМ) с помощью трехлучевой рабочей станции NVision 40 (Carl Zeiss), оснащенной энергодисперсионным микрозондовым анализатором Oxford Instruments X-MAX 80.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
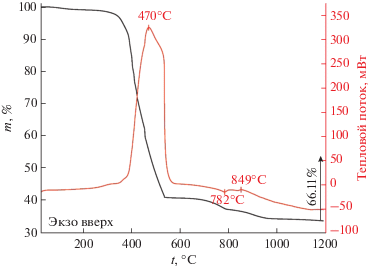
Термическое поведение порошка, полученного в результате гликоль-цитратного синтеза, было изучено с помощью синхронного ТГА/ДСК-анализа в потоке воздуха в интервале температур 25–1200°С. Как видно из термограммы (рис. 2), процесс нагрева порошка сопровождается пятью основными ступенями потери массы (Δm ~ 66.11%): 25–240 (Δm ~ 1%), 240–395 (~7.64%), 395–552 (~50.29%), 552–796 (~3.56%), 796–1200°С (~3.62%). Первые две ступени (общая потеря массы составляет ~8.64%) могут быть связаны с процессами десорбции воды и атмосферных газов с поверхности порошка. Третья, наиболее интенсивная ступень, сопровождаемая ярко выраженным экзотермическим эффектом с максимумом при 470°С, обусловлена процессами окисления остаточного углерода, образовавшегося в ходе синтеза при разложении органических компонентов. Потеря массы, сопровождаемая эндоэффектом с минимумом при ~782°С, согласно данным работы [35], вероятно, связана с процессами разложения промежуточных продуктов, обусловленных неполным протеканием взаимодействия реагентов в ходе синтеза или формированием карбонатов, в частности, карбоната с орторомбической структурой типа арагонита ar-Ba0.5Sr0.5CO3. Дальнейшее увеличение температуры выше 850°С приводит к постепенному формированию целевого оксида с кубической структурой типа перовскита, сопровождаемому незначительной потерей массы (Δm ~ 3.62%), которая, по данным авторов [36, 37], может быть вызвана изменением кислородной стехиометрии в структуре твердого раствора, а также трансформацией гексагональной кристаллической структуры Ba0.5Sr0.5Co0.8Fe0.2O3 – δ, существующей при более низких температурах, в кубическую. Таким образом, на основании данных термического анализа были определены условия дальнейшей термической обработки полученного порошка с целью формирования оксида заданного состава, не содержащего кристаллических примесей (1200°С, 1 ч).
Рис. 2.
Кривые ТГА (черная) и ДСК (красная) в потоке воздуха для порошка, полученного после воспламенения реакционной системы.
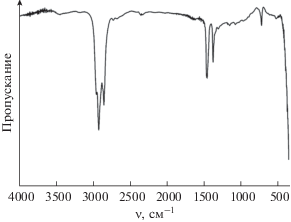
Согласно данным ИК-спектроскопии (рис. 3), порошок после термической обработки характеризуется наличием малоинтенсивных полос поглощения в областях 3100–3600 (валентные колебания OH-групп) и 1600–1700 см–1 (деформационные колебания НОН), свидетельствующих о незначительном количестве адсорбированной воды на его поверхности. Кроме того, отсутствие характеристических полос поглощения ${\text{CO}}_{{\text{3}}}^{{{\text{2}}--}}$-групп с максимумами при 857 и 1063 см–1 свидетельствует об отсутствии карбонатов (в частности, Sr0.6Ba0.4CO3, SrCO3 и Ba0.5Sr0.5CO3) в составе целевого оксида [38, 39]. Полосы поглощения в интервале 450–650 см–1, характерные для связей M–O, указывают на формирование оксида целевого состава [40, 41].
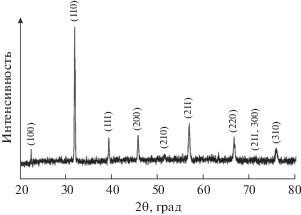
Результаты РФА (рис. 4) исследуемого порошка хорошо согласуются с данными, полученными с помощью ИК-спектроскопии, и подтверждают формирование твердого раствора заданного состава с кристаллической структурой типа перовскита (Pm$\bar {3}$m), о чем свидетельствуют рефлексы на рентгенограмме при 22°, 32°, 39°, 46°, 52°, 57°, 67°, 71°, 76°, соответствующие кристаллографическим плоскостям (100), (110), (111), (200), (210), (211), (220), (221, 300) и (310).
Микроструктура полученного оксидного порошка была исследована с помощью растровой электронной микроскопии (рис. 5). Как видно из микрофотографий, полученный порошок после термообработки характеризуется наличием спеченных агломератов со средним размером ~25 мкм. Изучение микроструктуры порошка в режиме контраста по среднему атомному номеру (детектор отраженных электронов) и результаты рентгеноспектрального элементного анализа свидетельствуют об однородном распределении элементов в структуре исследуемого твердого раствора и отсутствии примесных включений.

ЗАКЛЮЧЕНИЕ
Изучен процесс гликоль-цитратного синтеза оксида состава Ba0.5Sr0.5Co0.8Fe0.2O3 – δ. Согласно результатам РФА, ИК-спектроскопии, а также рентгеноспектрального элементного анализа в рамках растровой электронной микроскопии предложенный метод и условия синтеза, а также последующей термической обработки позволяют получать твердый раствор целевого состава, характеризующийся кубической кристаллической структурой типа перовскита (Pm$\bar {3}$m). Установлено, что полученный оксид не содержит кристаллических примесей, которые могли бы негативно сказаться на его функциональных характеристиках.
Исходя из экспериментальных данных, полученных с использованием комплекса физико-химических методов анализа (РФА, РЭМ, ИК-спектроскопии, синхронного ТГА/ДСК-анализа), синтезированный оксид по своим физико-химическим характеристикам и микроструктурным особенностям представляет практический интерес; на его основе в дальнейшем планируется получение дисперсных систем, подходящих по своим реологическим характеристикам для применения в качестве функциональных чернил при формировании катодных покрытий для среднетемпературных твердооксидных топливных элементов с помощью печатных технологий.
Список литературы
Michaelides E.E. Alternative Energy Sources. Berlin: Heidelberg, 2012. https://doi.org/10.1007/978-3-642-20951-2
Kalinina M.V., Morozova L.V., Khlamov I.I. et al. // Glass. Phys. Chem. 2014. V. 40. № 5. https://doi.org/10.1134/S108765961405006X
Faro M.L., Trocino S., Zignani S.C. et al. // Compend. Hydrog. Energy. Elsevier, 2016. P. 89. https://doi.org/10.1016/B978-1-78242-363-8.00004-9
Mahato N., Banerjee A., Gupta A. et al. // Prog. Mater. Sci. 2015. V. 72. P. 141. https://doi.org/10.1016/j.pmatsci.2015.01.001
Egorova T.L., Kalinina M.V., Simonenko E.P. et al. // Russ. J. Inorg. Chem. 2017. V. 62. № 10. P. 1275. https://doi.org/10.1134/S0036023617100072
Kalinina M.V., Morozova L.V., Egorova T.L. et al. // Glass. Phys. Chem. 2016. V. 42. № 5. P. 505. https://doi.org/10.1134/S1087659616050060
Simonenko T.L., Bocharova V.A., Gorobtsov P.Y. et al. // Russ. J. Inorg. Chem. 2020. V. 65. № 9. P. 1292. https://doi.org/10.1134/S0036023620090193
Nesaraj A.S. // J. Sci. Ind. Res (India). 2010. V. 69. № 3. P. 169.
Tarancón A. // Energies. 2009. V. 2. № 4. P. 1130. https://doi.org/10.3390/en20401130
Simonenko T.L., Simonenko N.P., Mokrushin A.S. et al. // Ceram. Int. 2020. V. 46. № 1. P. 121. https://doi.org/10.1016/j.ceramint.2019.08.241
Simonenko T.L., Ivanova V.M., Simonenko N.P. et al. // Russ. J. Inorg. Chem. 2019. V. 64. № 14. P. 1753. https://doi.org/10.1134/S0036023619140080
Xu X., Wang H., Fronzi M. et al. // J. Mater. Chem. A. 2019. V. 7. № 36. P. 20624. https://doi.org/10.1039/c9ta05300j
Mahadik P.S., Shirsat A.N., Saha B. et al. // J. Therm. Anal. Calorim. 2019. V. 137. № 6. P. 1857. https://doi.org/10.1007/s10973-019-08082-2
Choi J., Im J., Park I. et al. // Ceram. Int. 2012. V. 38. 1. P. S489. https://doi.org/10.1016/j.ceramint.2011.05.060
Shao Z., Halle S.M. // Nature. 2004. V. 431. № 7005. P. 170. https://doi.org/10.1038/nature02863
Kim J.S., Yeon D.H., Jung D.W. et al. // J. Power Sources. 2014. V. 249. P. 66. https://doi.org/10.1016/j.jpowsour.2013.10.053
Sameera Devi C., Prasad G. // Trans. Indian Ceram. Soc. 2018. V. 77. № 2. P. 67. https://doi.org/10.1080/0371750X.2018.1452633
Li Y., Wong L.M., Yu C.C. et al. // Surf. Coatings Technol. 2017. V. 320. P. 344. https://doi.org/10.1016/j.surfcoat.2016.12.051
Giuliano A., Carpanese M.P., Clematis D. et al. // J. Electrochem. Soc. 2017. V. 164. № 10. P. F3114. https://doi.org/10.1149/2.0161710jes
Shen F., Lu K. // Fuel Cells. 2018. V. 18. № 4. P. 457. https://doi.org/10.1002/fuce.201800044
Habiballah A.S., Osman N., Md Jani A.M. // Ceram. Int. 2020. V. 46. № 14. P. 23262. https://doi.org/10.1016/j.ceramint.2020.06.002
Shi H., Chu G., Tan W. et al. // Front. Chem. 2020. V. 7. P. 1. https://doi.org/10.3389/fchem.2019.00947
Hung I.-M., Laing C.-Y. // ECS Trans. 2019. V. 25. № 2. P. 2645. https://doi.org/10.1149/1.3205823
Yamaguchi Y., Sumi H., Shimada H. et al. // J. Ceram. Soc. Jpn. 2019. V. 127. № 7. P. 485. https://doi.org/10.2109/jcersj2.19013
Wei B., Lü Z., Huang X. et al. // J. Eur. Ceram. Soc. 2006. V. 26. № 13. P. 2827. https://doi.org/10.1016/j.jeurceramsoc.2005.06.047
Li S., Lü Z., Wei B. et al. // J. Alloys Compd. 2008. V. 448. № 1–2. P. 116. https://doi.org/10.1016/j.jallcom.2006.10.032
Toprak M.S., Darab M., Syvertsen G.E. et al. // Int. J. Hydrogen Energy. 2010. V. 35. № 17. P. 9448. https://doi.org/10.1016/j.ijhydene.2010.03.121
Liu P., Kong J., Liu Q. et al. // J. Solid State Electrochem. 2014. V. 18. № 6. P. 1513. https://doi.org/10.1007/s10008-013-2374-y
Mukhopadhyay J., Basu R.N. // ECS Trans. 2013. V. 57. № 1. P. 1945. https://doi.org/10.1149/05701.1945ecst
Zhao Z., Liu L., Zhang X. et al. // Int. J. Hydrogen Energy. 2012. V. 37. № 24. P. 19036. https://doi.org/10.1016/j.ijhydene.2012.09.142
Sun S., Cheng Z. // J. Electrochem. Soc. 2017. V. 164. № 2. P. F81. https://doi.org/10.1149/2.0611702jes
Qian J., Tao Z., Xiao J. et al. // Int. J. Hydrogen Energy. 2013. V. 38. № 5. P. 2407. https://doi.org/10.1016/j.ijhydene.2012.11.126
Mosiałek M., Dudek M., Michna A. et al. // J. Solid State Electrochem. 2014. V. 18. № 11. P. 3011. https://doi.org/10.1007/s10008-014-2457-4
Kautkar P.R., Shirbhate S.C., Acharya S.A. // AIP Conf. Proc. 2018. V. 1953. P. 8. https://doi.org/10.1063/1.5032904
Nuernberg R.B., Morelli M.R. // Ceram. Int. 2016. V. 42. № 3. P. 4204. https://doi.org/10.1016/j.ceramint.2015.11.094
Zeljković S., Miyawaki J., Vranković D. et al. // Process. Appl. Ceram. 2018. V. 12. № 4. P. 342. https://doi.org/10.2298/PAC1804342Z
Deganello F., Liotta L.F., Marcì G. et al. // Mater. Renew. Sustain. Energy. 2013. V. 2. № 1. https://doi.org/10.1007/s40243-013-0008-z
Zhang Y., Yang G., Chen G. et al. // ACS Appl. Mater. Interfaces. 2016. V. 8. № 5. P. 3003. https://doi.org/10.1021/acsami.5b09780
Tan K.H., Rahman H.A., Taib H. et al. // Key Eng. Mater. 2018. V. 791. P. 66. https://doi.org/10.4028/www.scientific.net/KEM.791.66
Bohlender C., Kahnes M., Müller R. et al. // J. Mater. Sci. 2019. V. 54. № 2. P. 1136. https://doi.org/10.1007/s10853-018-2916-x
Ahmad N., Alam M., Adil S.F. et al. // J. King Saud Univ. - Sci. 2020. V. 32. № 3. P. 2059. https://doi.org/10.1016/j.jksus.2020.02.015
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии