

Журнал неорганической химии, 2021, T. 66, № 4, стр. 464-469
Синтез CaCu3Ti4O12, исследование влияния термообработки на морфологию и диэлектрические свойства
К. В. Иванов a, *, А. В. Носков a, О. В. Алексеева a, А. В. Агафонов a
a Институт химии растворов им. Г.А. Крестова РАН
153045 Иваново, ул. Академическая, 1, Россия
* E-mail: ivk@isc-ras.ru
Поступила в редакцию 14.10.2020
После доработки 14.10.2020
Принята к публикации 19.10.2020
Аннотация
Методом жидкофазного синтеза в среде уксусной кислоты получен керамический порошок – предшественник CaCu3Ti4O12. Синтезированные образцы были прокалены при 200, 400, 600, 800 и 1100°С и исследованы физико-химическими методами анализа. Исследования показали, что увеличение температуры отжига приводит к изменениям гранулометрического состава порошка и количественных показателей пористости. Анализ диэлектрических спектров суспензий синтезированных материалов позволяет заключить, что высокотемпературный отжиг порошка ведет к уменьшению времени релаксации.
ВВЕДЕНИЕ
В последние годы внимание исследователей привлекают керамические материалы со структурой перовскита в связи с возможностью их применения в высокочастотной электронике для производства конденсаторов, генераторов, фильтров [1]. К таким материалам относятся титанаты бария, стронция и кальция, однако для этих перовскитов характерно скачкообразное изменение диэлектрической проницаемости вблизи температуры Кюри, что существенно ограничивает возможности их практического применения.
Среди перовскитоподобных оксидов можно выделить титанат кальция-меди CaCu3Ti4O12 (CCTO) и родственные ему материалы. Диэлектрическая проницаемость этого соединения мало изменяется в широком диапазоне температур вплоть до 600 K, что позволяет использовать CCTO для создания элементов микроэлектроники, а именно: в конденсаторах, СВЧ-устройствах, газовых сенсорах, электронных устройствах автомобиля, оперативно-запоминающих устройствах и т.д. [2, 3].
Разработка новых методов синтеза наноразмерного CaCu3Ti4O12 представляет большой интерес в связи с возможностью уменьшения размеров элементов электроники, увеличения их эффективности и производительности. Наиболее распространенным методом синтеза ССТО, как и большинства перовскитов, является твердофазный вследствие своей простоты [4–8]. Однако наряду с титанатом кальция-меди при таком синтезе может образовываться оксид меди(II), заполняющий межзеренное пространство керамики и оказывающий влияние на функциональные свойства материала. Кроме того, время термической обработки исходных компонентов смеси занимает более 1 ч, что приводит к существенным энергетическим затратам при производстве CCTO [9].
В литературе имеется описание и других методов получения CaCu3Ti4O12, среди которых можно выделить жидкофазный синтез (в том числе золь-гель), синтез горения, соосаждения и др. [10–16]. Преимуществом жидкофазных методов синтеза по сравнению с твердофазным является более низкая температура процесса, а также возможность получения порошков с точной стехиометрией и однородным фазовым составом. В зависимости от используемой методики жидкофазного синтеза можно регулировать не только размеры частиц, но и их форму, удельную площадь поверхности и структуру пор. Авторами работы [17] золь-гель методом с последующим отжигом при 900°C в течение 2 ч был получен порошок ССТО с частицами размером <100 нм.
В обзоре [10] приводятся данные о влиянии условий получения CaCu3Ti4O12 (температура отжига, продолжительность) и примесей на его диэлектрические свойства, а также продемонстрирована актуальность подобных исследований для решения как фундаментальных, так и прикладных задач.
Настоящая работа является продолжением более раннего исследования [18], в котором методами рентгенофазового анализа, масс-спектроскопии, термического анализа, УФ- и ИК-спектроскопии была изучена термическая эволюция предшественника керамического порошка ССТО и его фотокаталитическая активность. Было установлено, что формирование фазы CaCu3Ti4O12 завершается к 1100°С.
В представленной статье предшественник ССТО был синтезирован жидкофазным методом и продолжены исследования его физико-химических свойств в зависимости от температуры термообработки. В частности, были использованы методы динамического светорассеяния, сорбции-десорбции паров азота и диэлектрической спектроскопии.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Синтез предшественника керамического порошка ССТО был проведен жидкофазным методом в среде уксусной кислоты. В качестве исходных реагентов использовали Ca(OH)2, Cu(CH3COO)2 ⋅ H2O и Ti(C4H9O)4 (Sigma-Aldrich, США) в стехиометрическом соотношении CaO : CuO : TiO2 = 1 : 3 : 4. На первом этапе синтеза были приготовлены водный раствор Cu(CH3COO)2 ⋅ H2O и раствор Ca(OH)2 в уксусной кислоте. Затем полученные растворы смешивали друг с другом и перемешивали в течение 1 ч при 85°С. К полученной однородной смеси по каплям приливали Ti(C4H9O)4 и также выдерживали в течение 1 ч при температуре 85°С и непрерывном перемешивании. Далее раствор выпаривали в сушильном шкафу при 100°С до постоянной массы и полученный образец отжигали в воздушной среде при температурах 200, 400, 600, 800 и 1100°С.
Образцы термически обработанных порошков были исследованы с помощью ряда физико-химических методов.
Гранулометрический состав был определен с помощью лазерного дифракционного анализатора Zetasizer Nano ZS (Malvern Instruments, Великобритания) в диапазоне от 0.3 нм до 10 мкм. С целью разрушения агломератов порошки предварительно обрабатывали в ультразвуковой ванне в среде изопропилового спирта в течение 1 ч.
Удельную поверхность и количественные характеристики пористой структуры отожженных образцов синтезированного порошка оценивали на основе анализа низкотемпературных (77 K) изотерм адсорбции-десорбции паров азота. Изотермы регистрировали с помощью высокоскоростного газового сорбционного анализатора QuantaChrome Nova 1200 (США).
Измерения диэлектрических характеристик 30%-ных суспензий полученных материалов проводили с помощью RCL-метра Е7-20 (Беларусь) в интервале частот от 25 до 106 Гц в ячейке конденсаторного типа. Необходимые количества твердой фазы и силиконового масла ПМС-20 (компания ПЕНТА) тщательно растирали в агатовой ступке в течение 2 ч до получения однородной устойчивой суспензии. Использование в работе силиконового масла предполагает исследование его влияния на особенности поведения порошка предшественника ССТО в процессе термической обработки до CaCu3Ti4O12 на диэлектрические характеристики в жидкой фазе. Выбор силиконового масла (ПМС-20) обусловлен его высокими эксплуатационными характеристиками: широким рабочим температурным диапазоном (от –60 до +200°C) и высоким значением пробойного напряжения (>7 кВ/мм).
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Гранулометрический состав
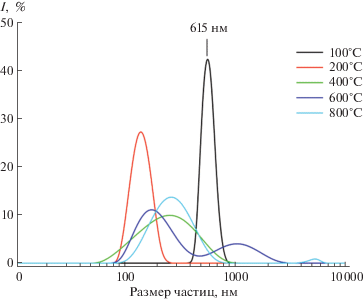
На рис. 1 представлены кривые распределения частиц исследованного порошка по размерам в зависимости от температуры отжига. Видно, что практически все частицы имеют поперечный размер в интервале от 100 до 2000 нм. Для порошка, отожженного при температуре 100°С, кривая распределения имеет мономодальный характер с максимумом при 615 нм. Увеличение температуры отжига приводит к увеличению доли более мелких частиц. Кроме того, в случае 600 и 800°С наблюдается бимодальный вид кривых распределения.
Пористая структура
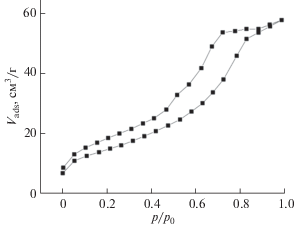
Исследования пористости отожженных образцов предшественников CaCu3Ti4O12, проведенные методом низкотемпературной адсорбции-десорбции азота, показали, что независимо от температуры отжига полученные изотермы относятся к IV типу согласно классификации IUPAC [19]. Наличие широкой петли гистерезиса (рис. 2) свидетельствует о микро- и мезопорах в структуре материала.
Для количественной оценки пористости исследуемых порошков полученные экспериментальные данные были проанализированы с использованием моделей Брунауэра–Эммета–Теллера (BET), Барретта–Джойнера–Халенды (BJH), Френкеля–Холси–Хилла (FHH) [20–25]. Расчеты проводили с учетом площади поперечного сечения молекулы N2 0.162 нм2, толщины монослоя N2 0.354 нм и плотности жидкого азота 0.808 г/см3.
Значения текстурных параметров, определенные из анализа изотерм для образцов предшественников CaCu3Ti4O12, отожженных при 200, 400, 600, 800 и 1100°C, представлены в табл. 1. Это следующие параметры:
Таблица 1.
Количественные характеристики пористости отожженных образцов предшественников CaCu3Ti4O12
| Параметр | Температура отжига, °С | ||||
|---|---|---|---|---|---|
| 200 | 400 | 600 | 800 | 1100 | |
| SBET, м2/г | 4.283 | 83.669 | 54.074 | 35.234 | 2.078 |
| SBJH, м2/г | 3.628 | 70.903 | 61.914 | 20.746 | 2.266 |
| VT, см3/г | 0.008 | 0.110 | 0.090 | 0.077 | 0.003 |
| Dav, нм | 7.162 | 5.305 | 6.633 | 8.697 | 6.174 |
| Dprob, нм | 3.984 | 3.571 | 4.016 | 3.144 | 4.012 |
SBET и SBJH – значения общей площади поверхности, рассчитанные с использованием моделей BET и BJH соответственно;
VT – общий объем пор, определяемый как объем жидкого азота, соответствующий его количеству, адсорбированному при относительном давлении p/p0 = 0.99;
Dav – средний диаметр пор;
Dprob – наиболее вероятный диаметр пор, соответствующий максимуму распределения пор по размерам.
Как видно из табл. 1, для образцов, отожженных при 400°С, объем порового пространства более чем в 10 раз превосходит соответствующее значение для образцов, отожженных при 200°С. Этот результат согласуется с данными термического анализа и масс-спектрометрии, представленными ранее в работе [19], из которых следует, что нагрев синтезированного порошка до 320°С сопровождается удалением воды, ацетатных групп и ацетона. Поэтому можно предположить, что благодаря этому удалению освобождается значительный объем порового пространства, доступный для азота.
Удельная поверхность отожженных образцов была определена с использованием моделей BET и BJH. Как видно из табл. 1, с увеличением температуры отжига выше 400°С значения SBET и SBJH уменьшаются. Также можно видеть некоторое различие в полученных значениях. Эта разница, по-видимому, связана с тем, что модели BET и BJH основаны на разных предположениях. Например, модель BJH основана на теории Кельвина о заполнении цилиндрических пор, тогда как в теории BET это допущение отсутствует.
На рис. 3 приведены диаграммы, характеризующие распределение пор по размерам. Они отражают доли (в процентах) общего порового пространства (V1 и V2), соответствующие малым (<11 нм в диаметре) и большим (>11 нм) порам. Видно, что почти для всех отожженных образцов (за исключением 800°С) бóльшая часть азота адсорбирована в порах диаметром <11 нм. При этом средний размер пор принимает значения в интервале от 5.3 до 8.7 нм.
Рис. 3.
Распределение порового пространства в зависимости от размеров пор при различных температурах отжига синтезированного порошка.
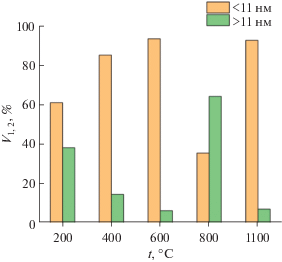
Данные, представленные на рис. 3, свидетельствуют о том, что в процессе термической обработки предшественника ССТО наблюдается нелинейное изменение размеров пор. Для пор диаметром <11 нм эти изменения обусловлены протеканием ряда физико-химических процессов, а именно удалением физически и химически связанной воды при температурах 100–250°C, а также избытка уксусной кислоты и продуктов термической деструкции органических включений исходных компонентов при нагревании до 600°C. Уменьшение доли таких пор (V1) при 800°C связано, по-видимому, с процессами спекания. Дальнейшее увеличение порового пространства, образованного этими порами, при 1100°C, скорее всего, вызвано фазовыми изменениями, происходящими в материале, в результате которых полностью заканчивается формирование ССТО.
Для больших пор (диаметром >11 нм) с ростом температуры также наблюдается неоднородное изменение. До 600°C происходит уменьшение порового пространства вследствие процессов спекания. Однако при 800°C наблюдается обратный эффект, связанный, по-видимому, с удалением при этой температуре карбонатных групп, образующихся вследствие взаимодействия образца с CO2 из окружающей среды. Соответственно, дальнейший отжиг при 1100°C вновь приводит к спеканию с последующим уменьшением доли больших пор (V2).
Количественная оценка неоднородности исследованных материалов была выполнена с использованием фрактальной модели FHH [24, 25], а именно – данные низкотемпературной адсорбции-десорбции паров N2 были использованы для определения фрактальной размерности поверхности пористого материала df.
Согласно [24], метод FHH позволяет рассчитать значение фрактальной размерности поверхности адсорбента с учетом эффектов поверхностного натяжения адсорбата, когда поверхностное натяжение жидкость/газ (капиллярная сила) стремится к уменьшению площади поверхности раздела. Для этого случая аналитическое выражение изотермы FHH имеет вид:
(1)
$\frac{{{{V}_{{{\text{ads}}}}}}}{{{{V}_{m}}}} \approx {{\left[ {RT\lg \left( {\frac{{{{p}_{{\text{0}}}}}}{p}} \right)} \right]}^{{{{d}_{f}} - 3}}},$Таблица 2.
Значения фрактальной размерности поверхности предшественника ССТО в зависимости от температуры отжига, найденные методом FHH
| Температура отжига, °С | df | |
|---|---|---|
| адсорбция | десорбция | |
| 200 | 2.637 | 2.713 |
| 400 | 2.661 | 2.695 |
| 600 | 2.645 | 2.659 |
| 800 | 2.623 | 2.627 |
Как видно из табл. 2, наблюдается уменьшение фрактальной размерности по мере увеличения температуры отжига начиная с 400°С. Это свидетельствует о снижении степени развитости поверхности и согласуется с установленными методами BET и BJH тенденциями снижения удельной поверхности и порового пространства.
Диэлектрические измерения
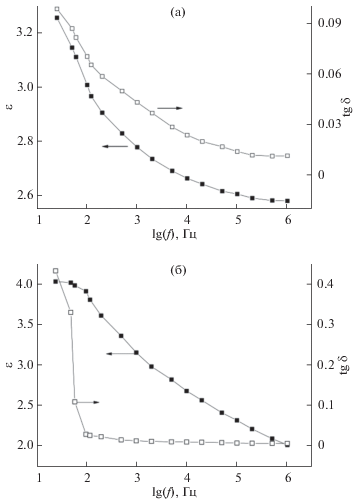
В настоящей работе было изучено влияние термообработки на частотные зависимости диэлектрической проницаемости и тангенса угла диэлектрических потерь суспензий синтезированных порошков в силиконовом масле. Полученные результаты представлены на рис. 4. Как видно из рисунка, в частотном интервале 25–106 Гц наблюдается дисперсия диэлектрической проницаемости. При этом значения tg δ не превышают 0.1 для необработанного порошка (рис. 4а). В случае высокой температуры отжига (1100°С) значения tg δ не превышают 0.03 при частотах в интервале 100–106 Гц (рис. 4б).
Рис. 4.
Частотные зависимости диэлектрической проницаемости и тангенса угла диэлектрических потерь для суспензий синтезированных порошков: без термообработки (а); отожженных при 1100°С (б).
В соответствии с теорией Дебая, аналитическая зависимость диэлектрической проницаемости от циклической частоты переменного тока (ω = 2πf) имеет вид:
(2)
$\varepsilon = {{\varepsilon }_{\infty }} + \frac{{{{\varepsilon }_{{{\text{st}}}}} - {{\varepsilon }_{\infty }}}}{{1 + {{{\left( {\omega \tau } \right)}}^{2}}}},$В настоящей работе уравнение (2) было использовано для математической обработки полученных экспериментальных зависимостей диэлектрической проницаемости от частоты, что позволило рассчитать время релаксации исследованных суспензий синтезированных порошков. Было установлено, что отжиг порошка ведет к уменьшению времени релаксации: если в случае необработанного порошка значение τ составляет 3.1 × 10–4 с, то для отожженного при 1100°С – τ = = 3.5 × 10–5 с.
ЗАКЛЮЧЕНИЕ
Проведенные исследования показали, что полученный в процессе жидкофазного синтеза предшественник керамического порошка ССТО претерпевает значительные изменения при термической обработке. В частности, наблюдаются изменения гранулометрического состава порошка, количественных характеристик пористой структуры, параметров диэлектрической релаксации. По-видимому, выявленная термическая эволюция связана с формированием фазы CaCu3Ti4O12, завершающимся, по данным РФА, к 1100°С.
Список литературы
Raia A.K., Mandala K.D., Kumar D. et al. // J. Mater. Chem. Phys. 2010. V. 122. № 1. P. 217. https://doi.org/10.1016/j.matchemphys.2010.02.037
Liu P., Lai Y., Zeng Y. et al. // J. Alloys Compd. 2015. V. 650. P. 59. https://doi.org/10.1016/j.jallcom.2015.07.247
Homes C.C., Vogt T., Shapiro et al. // J. Science. 2001. V. 293. № 5530. P. 673. https://doi.org/10.1126/science.1061655
Wang B., Pu Y.P., Wua H.D. et al. // Ceram. Int. 2013. V. 39. № 1. P. 525. https://doi.org/10.1016/j.ceramint.2012.10.127
Tang Z., Huang Y., Wu K. et al. // J. Eur. Ceram. Soc. 2018. V. 38. P. 1569. https://doi.org/10.1016/j.jeurceramsoc.2017.11.018
Schmidt R., Stennett M.C., Hyatt N.C. et al. // J. Eur. Ceram. Soc. 2012. V. 32. P. 3313. https://doi.org/10.1016/j.jeurceramsoc.2012.03.040
Almeida-Didry S.D., Merad S., Autret-Lambert C. et al. // Solid State Sci. 2020. V. 109. https://doi.org/10.1016/j.solidstatesciences.2020.106431
Almeida-Didry S.D., Autret C., Honstettre C. et al. // Solid State Sci. 2016. V. 61. P. 102. https://doi.org/10.1016/j.solidstatesciences.2016.07.010
Sekushin N.A., Zhuk N.A., Koksharova L.A. et al. // Lett. Mater. V. 9. № 1. P. 5. https://doi.org/10.22226/2410-3535-2019-1-5-10
Ahmadipour M., Ain M.F., Ahmad Z.A. // J. Nano-Micro Lett. 2016. V. 8. № 4. P. 291. https://doi.org/10.1007/s40820-016-0089-1
Yuan W.X., Hark S.K., Mei W.N. et al. // J. Electrochem. Soc. 2010. V. 157. № 5. P. 117. https://doi.org/10.1149/1.3353040
Hua W.M., Fu Z., Li W.Q. et al. // J. Cent. South Univ. 2012. V. 19. № 12. P. 3385. https://doi.org/10.1007/s11771-012-1418-2
Banerjee N., Krupanidhi S.B. // Curr. Nanosci. 2010. V. 6. № 4. P. 432. https://doi.org/10.2174/157341310791658955
Kim K.M., Kim S.J., Lee J.H. et al. // J. Eur. Ceram. Soc. 2007. V. 27. № 13. P. 3991. https://doi.org/10.1016/j.jeurceramsoc.2007.02.081
Agafonov A.V., Ivanov K.V., Davydova O.I. et al. // Russ. J. Inorg. Chem. 2011. V. 56. № 7. P. 1025. [Агафонов А.В., Иванов К.В., Давыдова О.И. и др. // Журн. неорган. химии. 2011. Т. 56. № 7. С. 1087.]https://doi.org/10.1134/S0036023611070035
Zhao J., Liu J., Ma G. // Ceram. Int. 2012. V. 38. P. 1221. https://doi.org/10.1016/j.ceramint.2011.08.052
Zhao J., Liu J., Ma G. // J. Ceram. Int. 2012. V. 38. № 2. P. 1221. https://doi.org/10.1016/j.ceramint.2011.08.052
Ivanov K.V., Alekseeva O.V., Agafonov A.V. // Russ. J. Inorg. Chem. 2020. V. 65. № 10. P. 1338. [Иванов К.В., Алексеева О.В., Агафонов А.В. // Журн. неорган. химии. 2020. Т. 65. № 10. С. 1338. https://doi.org/10.31857/s0044457x20100098]https://doi.org/10.1134/S0036023620100095
Sing K.S.W., Everett D.H., Haul R.A.W. et al. // Pure Appl. Chem. 1985. V. 57. P. 603. https://doi.org/10.1515/iupac.57.0007
Barrett E.P., Joyner L.G., Halenda P.P. // J. Am. Chem. Soc. 1951. V. 73. P. 373. https://doi.org/10.1021/ja01145a126
Sing K.S.W. // Adv. Coll. Interf. Sci. 1998. V. 76–77. P. 3. https://doi.org/10.1016/s0001-8686(98)00038-4
Aligizaki K.K. Pore Structure of Cement-Based Materials: Testing Interpretation and Requirements (Modern Concrete Technology). N.Y.: Taylor & Francis, 2005. https://doi.org/10.1201/9781482271959
Panella V., Krim J. // Studies in Surface Science and Catalysis. 1994. V. 87. P. 91. https://doi.org/10.1016/s0167-2991(08)63068-2
Pomoni P.J., Tsaous E.T. // Langmuir. 2009. V. 25. P. 9986. https://doi.org/10.1021/la901121c
Sandoval-Díaz L.-E., Aragon-Quiroz J.-A., Ruíz-Cardona Y.-S. et al. // Microporous and mesoporous materials. 2017. V. 237. P. 260. https://doi.org/10.1016/j.micromeso.2016.08.030
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии