

Журнал неорганической химии, 2021, T. 66, № 4, стр. 477-484
Комплексы Ru(III) типа NAMI-A с лигандами на основе лонидамина и бексаротена как антипролиферативные агенты
И. А. Шутков a, А. А. Антонец a, В. Ю. Тюрин a, Е. Р. Милаева a, А. А. Назаров a, *
a Московский государственный университет им. М.В. Ломоносова
119991 Москва, Ленинские горы, 1, Россия
* E-mail: nazarov@med.chem.msu.ru
Поступила в редакцию 11.08.2020
После доработки 09.10.2020
Принята к публикации 12.10.2020
Аннотация
В настоящее время активно ведется поиск новых металлосодержащих противоопухолевых агентов. Хотя препараты платины успешно применяются в клинической практике, они не лишены недостатков, таких как высокая общая токсичность и первичная или приобретенная резистентность. Наилучший потенциал среди неплатиновых противоопухолевых агентов показали соединения рутения, два из которых, NAMI-A и KP1019 (КР1339), проходили или проходят клинические испытания. В данной работе представлен синтез полных аналогов соединения-лидера NAMI-A, содержащих фрагменты биологически активных органических молекул, а именно лонидамин и бексаротен. Изучено электрохимическое поведение комплексов Ru(III) и показана легкость восстановления, которая указывает на перспективность их использования в качестве противоопухолевых соединений, механизм действия которых, вероятно, основан на активации реакцией восстановления атома металла. Цитотоксичность полученных комплексов изучена in vitro, определены значения IC50. Выбраны два соединения, перспективные для дальнейшего изучения.
ВВЕДЕНИЕ
Введение в клиническую практику цисплатина привело к развитию нового направления в поиске противоопухолевых агентов [1–7]. Препараты платины успешно применяются в клинике и используются почти в половине всех химиотерапевтических схем [8, 9]. В настоящее время ведутся исследования по поиску новых противоопухолевых металлосодержащих соединений, лишенных таких недостатков цисплатина и его аналогов, как низкая избирательность и высокая токсичность, а также наличие первичной или приобретенной резистентности. Одной из основных стратегий поиска новых противоопухолевых соединений металлов является получение комплексов рутения, золота, осмия, галлия, титана, родия и др., которые будут обладать селективностью и отличным от платиновых лекарственных средств механизмом действия [10–13].
В ряду новых комплексов наиболее перспективными являются соединения рутения, два из которых, NAMI-A и KP1019 (КР1339 – натриевая соль соединения KP1019) (рис. 1), проходили или проходят клинические испытания [14–20]. Основное отличие противоопухолевых комплексов рутения от классических соединений платины – это отсутствие кросс-резистентности к платиновым препаратам, низкий уровень общей токсичности и белковые молекулярные мишени [21, 22].
Рис. 1.
Комплексы Ru(III), находящиеся в клинических испытаниях в качестве противоопухолевых агентов.
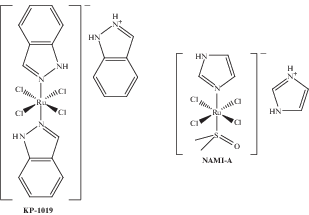
В доклинических исследованиях препарат NAMI-A на основе Ru(III) показал низкую активность по отношению к основной опухоли, но оказался эффективным при воздействии на метастазы. В I стадии клинических испытаний NAMI-A способствовал остановке прогрессирующего метастазирующего рака легких у тяжелобольного пациента на 21 неделю. Установлено, что соединение обладает низким уровнем общей токсичности [15].
Механизм действия NAMI-A до сих пор до конца не ясен. Существуют две основные гипотезы: первая заключается в связывании с биологическими мишенями, например с молекулой ДНК, по аналогии с соединениями платины; показана также возможность связывания продуктов гидролиза NAMI-A с РНК. Второй подход заключается во внутриклеточном восстановлении до активной формы – комплекса Ru(II), чем и объясняется низкий уровень общей токсичности по сравнению с цисплатином [19, 23–26].
В отличие от NAMI-A, комплекс KP1019 индуцирует апоптоз опухолевых клеток [27, 28], блокирует электронно-транспортную цепь и деполяризует мембрану митохондрий, вызывает активацию каспазы-3 и нарушение функции антиапоптотического фактора bcl-2 [29]. Статус апоптотического белка p53 опухолевых клеток свидетельствует о том, что действие на ДНК не является основным механизмом, однако есть вероятность нарушения структуры ДНК образующимися активными метаболитами кислорода. Соединение KP1019 оказалось активным по отношению ко многим хеморезистентным видам рака [29, 30].
Одним из современных подходов к увеличению специфичности препаратов в неорганической медицинской химии является создание соединений двойного действия, когда с токсичным фрагментом металла связана биологически активная молекула, которая селективно взаимодействует с молекулярными мишенями и обеспечивает действие на раковую клетку. Данный подход дает возможность одновременного влияния на несколько молекулярных особенностей злокачественных клеток с целью увеличения биологического ответа и эффективности соединений [31–43].
Ранее было показано, что введение биологически активных молекул в координационную сферу комплексов Ru(III) значительно влияет на противораковую активность. На основе этакриновой кислоты (ингибитор глутатион-S-трансферазы) получен аналог NAMI-A (рис. 2, 1), который проявляет меньшую цитотоксичность по сравнению с производным Ru(II), что характерно для комплексов Ru(III) [39, 44–46]. Модификацией бидентатного лиганда этилендиамина и имидазола в структуру был введен остаток гефитиниба (рис. 2, 2–3), который является ингибитором эпидермиального фактора роста (ЭФР). Наибольшую активность в ингибировании ЭФР проявил комплекс с имидазольным фрагментом, а комплекс с этилендиамином в присутствии эндогенного ЭФР оказался более активным, чем металлоорганические соединения Ru(II) [47, 48].
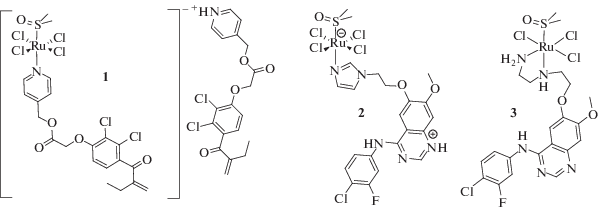
Исследования показывают, что в составе комплекса органический биологически активный фрагмент сохраняет свою активность и возможность связываться с молекулярной мишенью. Следовательно, данный подход предоставляет возможность создания мультитаргетных противоопухолевых соединений.
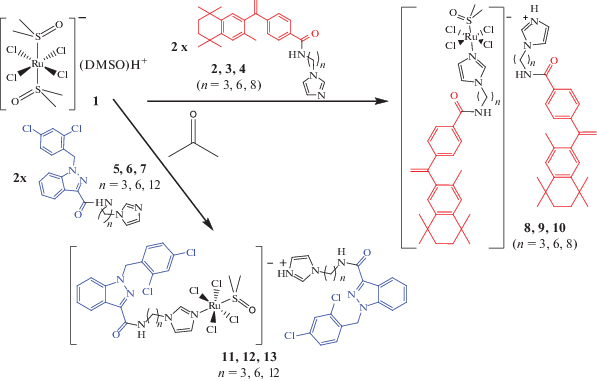
В данном сообщении мы представляем синтетический подход к получению координационных соединений Ru(III) – аналогов NAMI-A (рис. 3) с биологически активными лигандами на основе бексаротена и лонидамина, изучение их электрохимического поведения и антипролиферативной активности.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Растворители были очищены и дегазированы перед использованием. Элементный анализ проводили в лаборатории биоэлементоорганической химии химического факультета МГУ имени М.В. Ломоносова на анализаторе MicroCube Elementar. Масс-спектры ИЭР получали на спектрометре Bruker LC/MS – Ion-trap amaZon SL.
[Ru(ДМСО)(N-(3-(1Н-имидазол-1-ил)пропил)-4-(1-(3,5,5,8,8-пентаметил-5,6,7,8-тетрагидронафтален-2-ил)винил)бензамид)Cl4][(N-(3-(1Н-имидазолиум-1-ил)пропил)-4-(1-(3,5,5,8,8-пентаметил-5,6,7,8-тетрагидронафтален-2-ил)винил)бензамид)] (8)
Соединение 2 (61 мг, 0.1330 ммоль) в ~2 мл ацетона добавляли к раствору комплекса [Ru(DMSO)2Cl4]–(DMSO)2H+ (37 мг, 0.0665 ммоль) в ацетоне (8.0 мл). Реакционную смесь перемешивали в течение 12 ч. Раствор фильтровали, растворитель упаривали в вакууме, полученный оранжевый порошок высушивали в вакууме. Выход 42 мг (51.2%), tразл > 250°С. МС-ИЭР (m/z): 777 [M – HL+]–, 1232 [M – H+], 456 [HL]+.
[Ru(ДМСО)(N-(3-(1Н-имидазол-1-ил)гексил)-4-(1-(3,5,5,8,8-пентаметил-5,6,7,8-тетрагидронафтален-2-ил)винил)бензамид)Cl4][(N-(3-(1Н-имидазолиум-1-ил)гексил)-4-(1-(3,5,5,8,8-пентаметил-5,6,7,8-тетрагидронафтален-2-ил)винил)бензамид)] (9)
Соединение 9 получали аналогично 8 из лиганда 3 (66 мг, 0.1330 ммоль) и комплекса [Ru(DMSO)2Cl4]–(DMSO)2H+ (37 мг, 0.0665 ммоль) в ацетоне (10.0 мл). Выход 46 мг (52.5%), tразл > > 250°С. МС-ИЭР (m/z): 817 [M – HL+]–, 1316 [M – H+], 498 [HL]+.
[Ru(ДМСО)(N-(3-(1Н-имидазол-1-ил)октил)-4-(1-(3,5,5,8,8-пентаметил-5,6,7,8-тетрагидронафтален-2-ил)винил)бензамид)Cl4][(N-(3-(1Н-имидазолиум-1-ил)октил)-4-(1-(3,5,5,8,8-пентаметил-5,6,7,8-тетрагидронафтален-2-ил)винил)бензамид)] (10)
Соединение 10 получали аналогично 8 из лиганда 4 (120 мг, 0.2282 ммоль) и комплекса [Ru(DMSO)2Cl4]–(DMSO)2H+ (64 мг, 0.1141 ммоль) в ацетоне (12.0 мл). Выход 118 мг (75.3%), tразл > > 250°С. МС-ИЭР (m/z): 845 [M – HL+]–, 1372 [M – H+], 526 [HL]+.
[Ru(ДМСО)(N-(2-(1Н-имидазол-1-ил)пропил)-1-(2,4-дихлорбензил)-1Н-индазол-3-карбоксамид)Cl4][(N-(2-(1Н-имидазоиум-1-ил)пропил)-1-(2,4-дихлорбензил)-1Н-индазол-3-карбоксамид)] (11)
Соединение 11 получали аналогично 8 из лиганда 5 (100 мг, 0.2335 ммоль) и комплекса [Ru(DMSO)2Cl4]–(DMSO)2H+ (65 мг, 0.1167 ммоль) в ацетоне (12.0 мл). Выход 89 мг (73.5%), tразл > > 250°С. МС-ИЭР (m/z): 749 [M – HL+]–, 1178 [M – H+], 428 [HL]+.
[Ru(ДМСО)(N-(2-(1Н-имидазол-1-ил)гексил)-1-(2,4-дихлорбензил)-1Н-индазол-3-карбоксамид)Cl4][(N-(2-(1Н-имидазоиум-1-ил)гексил)-1-(2,4-дихлорбензил)-1Н-индазол-3-карбоксамид)] (12)
Соединение 12 получали аналогично 8 из лиганда 6 (150 мг, 0.3190 ммоль) и комплекса [Ru(DMSO)2Cl4]–(DMSO)2H+ (89 мг, 0.1595 ммоль) в ацетоне (17.0 мл). Выход 111 мг (62.1%), tразл > > 250°С. МС-ИЭР (m/z): 791 [M – HL+]–, 1262 [M – H+], 470 [HL]+.
[Ru(ДМСО)(N-(2-(1Н-имидазол-1-ил)додецил)-1-(2,4-дихлорбензил)-1Н-индазол-3-карбоксамид)Cl4][(N-(2-(1Н-имидазоиум-1-ил)додецил)-1-(2,4-дихлорбензил)-1Н-индазол-3-карбоксамид)] (13)
Соединение 13 получали аналогично 8 из лиганда 7 (81 мг, 0.1460 ммоль) и комплекса [Ru(DMSO)2Cl4]–(DMSO)2H+ (41 мг, 0.0730 ммоль) в ацетоне (10.0 мл). Выход 81 мг (86.1%), tразл > > 250°С. МС-ИЭР (m/z): 875 [M – HL+]–, 1430 [M – H+], 554 [HL]+.
Электрохимические исследования
Приборы и электроды. Регистрацию циклических вольтамперограмм (ЦВА) проводили с помощью цифрового потенциостата-гальваностата IPC-Pro М. Вольтамперограммы регистрировали на фоне 0.5 М n-Bu4NBF4 в CH3CN при 20°С в трехэлектродной ячейке K0264 MICRO-CELL. Рабочими электродами служили платиновый и стеклоуглеродный, электродом сравнения – хлорсеребряный электрод в насыщенном растворе KCl, вспомогательный электрод платиновый. Ацетонитрил марки “ч.” предварительно перегоняли, собирая фракцию с tкип = 81–82°С/760 мм рт. ст. Концентрация растворов исследуемых соединений составляла 10–3 моль/л. Кислород из ячейки удаляли продуванием сухого аргона. Все образцы вносили в раствор после полного удаления кислорода из ячейки. Число переносимых электронов определяли путем сравнения с высотой пика окисления ферроцена.
Определение цитотоксичности с использованием МТТ-теста
Клетки культивировали в стандартной среде DMEM (Gibco™), содержащей 5%-ную эмбриональную сыворотку телят (Gibco™) при 37°С и в 5%-ном СО2. Для определения цитотоксичности клетки рассевали в 96-луночные планшеты (“Eppendorf”, Германия) (7 × 103 клеток в 100 мкл культуральной среды) и инкубировали 24 ч. В день экспериментов готовили серийные разведения комплекса в диметилсульфоксиде (ДМСО) и вносили полученные растворы в культуру клеток в концентрациях 100, 50, 25, 12.5, 6.25, 3.12, 1.56, 0.78 мкМ. Далее клетки инкубировали в течение 72 ч, затем добавляли 10 мкл раствора реагента МТТ (5 мг/мл) в культуральной среде и инкубировали 50 мин при 37 град/СО2 5% для образования формазана. Среду удаляли, добавляли 100 мкл ДМСО и перемешивали. После полного растворения кристаллов формазана измеряли оптическую плотность содержимого лунок на мультилуночном спектрофотометре Zenith 200 rt при длине волны 570 нм. Данные представляли в виде оптической плотности экспериментальных образцов относительно контроля. За 100% принимали оптическую плотность в контроле, где клетки инкубировали в отсутствие комплекса, но в присутствии растворителя (ДМСО).
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Новые соединения Ru(III) получены замещением молекул ДМСО в комплексе Ru 1 на имидазольные лиганды, при этом один лиганд координируется, замещая ДМСО во внутренней сфере комплекса, а второй в результате протонирования группы имидазола замещает протонированную молекулу ДМСО во внешней сфере комплекса. Используя полученные ранее модифицированные имидазольные лиганды с различной длиной линкера [49, 50], которые обладают собственными молекулярными мишенями, получили новые полные аналоги соединения-лидера NAMI-A, в которых содержатся два биологически активных органических фрагмента и рутениевый центр (рис. 3). Комплексы выделяли в виде оранжевых порошков, состав полученных соединений подтверждали данными элементного анализа и методом масс-спектрометрии ИЭР.
В масс-спектрах соединений в режиме регистрации отрицательных ионов наблюдаются два сигнала с молекулярной массой, соответствующей предполагаемому аниону и недиссоциированному депротонированному комплексу с характерным изотопным распределением рутения, а в режиме регистрации положительно заряженных ионов наблюдается сигнал, который можно отнести к протонированному имидазольному лиганду (рис. 4). Наблюдаемые сигналы полностью соответствуют расчетным по массе и изотопному распределению.
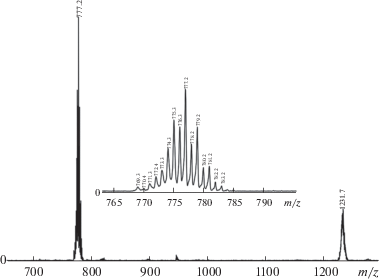
С целью оценить возможность внутриклеточного восстановления была проведена оценка редокс-активности новых комплексов Ru(III). Электрохимическое поведение соединений 8–10 и 11–13 в ацетонитриле было изучено методом циклической вольтамперометрии с использованием платинового и стеклоуглеродного (СУ) рабочих электродов. Показано, что лиганды не проявляют электрохимической активности, а для всех синтезированных комплексов наблюдаются редокс-переходы в анодной и катодной областях. Потенциалы переходов соединений 8–10 и 11–13 приведены в табл. 1 и 2.
Таблица 1.
Редокс-потенциалы комплексов 8–10 (скорость развертки 200 мВ/с, С = 10–3 моль/л, Ag/AgCl, фоновый электролит 0.5 М n-Bu4NBF4, Pt-электрод, CH3CN)
| Соединение | E red, В | $E_{1}^{{{\text{ox}}}}$, В | $E_{2}^{{{\text{ox}}}}$, В | $E_{3}^{{{\text{ox}}}}$, В |
|---|---|---|---|---|
| 8 | –0.65 (необратимо) | 0.09/0.1 | 1.28/1.17 | – |
| 9 | –0.82 (необратимо) | – | 1.27/1.2 | 1.77 |
| 10 | –0.59 (необратимо) | 0.17/0.1 | 1.280/1.19 | 1.9 |
Таблица 2.
Редокс-потенциалы комплексов 11–13, определенные с помощью стеклоуглеродного (СУ) и Pt-рабочих электродов (скорость развертки 200 мВ/с, С = 10–3 моль/л, фоновый электролит 0.5 М n-Bu4NBF4, Ag/AgCl)
| Соединение | СУ-электрод | Pt-электрод | ||||
|---|---|---|---|---|---|---|
| $E_{1}^{{{\text{red}}}}$, В | $E_{1}^{{{\text{ox}}}}$, В | $E_{2}^{{{\text{ox}}}}$, В | $E_{1}^{{{\text{red}}}}$, В | $E_{1}^{{{\text{ox}}}}$, В | $E_{2}^{{{\text{ox}}}}$, В | |
| 11 | –0.27/–0.43 | 0.19/0.11 | 1.27/1.18 | –0.12/–0.24 | 0.33/0.04 | 1.48/1.17* |
| 12 | –0.29/–0.43 | 0.17/0.10 | 1.26/1.17 | –0.8/–0.32 | 0.26/0.10 | 1.33/1.18* |
| 13 | –0.33/–0.48 | 0.27/0,10 | 1.28/1.13 | 0.46/0.06 | 1.53/1.21* | |
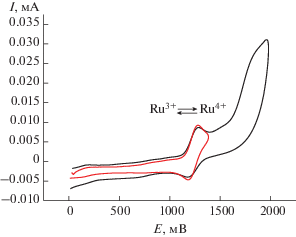
Электрохимическое поведение комплексов 8–13 во многом аналогично. В анодной области на платиновом электроде регистрируются одноэлектронные пики диффузионного характера в интервале 1.17–1.28 В для соединений 8–10 и 1.18–1.53 В для 11–13 и в диапазоне 1.17–1.28 В для комплексов 11–13 на СУ-электроде. В случае соединений 8–10 на Pt- и 11–13 на СУ-электродах пики на ЦВА имеют обратимый характер (рис. 5). Очевидно, данные переходы отвечают окислению Ru(III) в Ru(IV):
Рис. 5.
ЦВА соединения 10 на платиновом электроде (С = 10–3 моль/л, скорость развертки 200 мВ/с, Ag/AgCl, фон 0.5 М n-Bu4NBF4, CH3CN).
Кроме того, в анодной области ЦВА для всех полученных соединений наблюдаются квазиобратимые пики низкой интенсивности в области от 40 до 460 мВ (рис. 6). Согласно данным [51], этот пик может быть обусловлен редокс-переходом Ru(II) в Ru(III) c измененным лигандным окружением (возможна замена одного или двух атомов хлора на молекулу растворителя).
Рис. 6.
ЦВА комплекса 11 на СУ-электроде (С = = 10‒3 моль/л, скорость развертки 200 мВ/с, Ag/AgCl, фон 0.5 М n-Bu4NBF4, CH3CN).
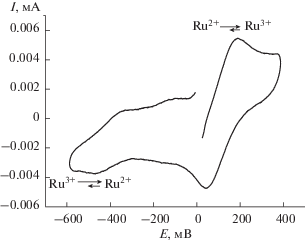
В катодной области потенциалов для соединений 11–13 наблюдаются одноэлектронные пики в интервале от –80 до –310 мВ при измерениях на платиновом электроде и от –135 до –410 мВ при измерениях на СУ-электроде соответственно. Данные значения потенциала, согласно литературным данным, соответствуют процессу восстановления Ru(III) в Ru(II) [50–53]. Квазиобратимый характер перехода (разница потенциалов между катодным и анодным пиками составляет ΔE = 150–260 мВ) свидетельствует об изменении геометрии комплексов. В случае комплексов 8–10 восстановление протекает необратимо при более отрицательных потенциалах (в интервале от –0.59 до –0.82 В), что свидетельствует о протекании быстрой химической стадии, следующей за переносом электрона.
На основании данных, полученных методом ЦВА, можно сделать вывод, что длина углеводородного спейсера в лиганде незначительно сказывается на значениях редокс-потенциалов. Легкость восстановления комплексов указывает на перспективность их использования в качестве противоопухолевых соединений, механизм действия которых основан на активации реакцией восстановления атома металла.
Оценка антипролиферативной активности изучена методом МТТ в экспериментах in vitro. Для соединений 8–13 определена концентрация полумаксимального ингибирования (IC50) на клетках аденокарциномы толстой кишки человека SW480, аденокарциномы молочной железы человека MCF7, аденокарциномы легкого человека А549 и рака толстой кишки человека HCT116 (табл. 3). Оценку цитотоксичности проводили при сравнении с исходными органическими соединениями: бексаротеном, лонидамином, а также с клинически используемым цисплатином.
Таблица 3.
Значения цитотоксичности для комплексов 8–10 и 11–13 – аналогов NAMI-A
| Соединение | Линкер, n | IC50, мкМ | |||
|---|---|---|---|---|---|
| A549 | HCT116 | MCF7 | SW480 | ||
| Цисплатин | 20.1 ± 1.9* | 9.7 ± 4.5 | 15.2 ± 5* | 17.6 ± 5* | |
| Бексаротен | 85 ± 9* | – | 67 ± 13* | 80 ± 10* | |
| 8 | 3 | 0.5 ± 0.1 | 27.1 ± 1.1 | 40.1 ± 6.1 | 52.8 ± 4.4 |
| 9 | 6 | >100 | >100 | >100 | >100 |
| 10 | 8 | >100 | >100 | >100 | >100 |
| Лонидамин | >90* | – | 30 ± 10* | >90* | |
| 11 | 3 | 78.6 ± 4.7 | >100 | 39.9 ± 1.6 | 52.5 ± 9.8 |
| 12 | 6 | 67.4 ± 1.9 | 73.9 ± 10.9 | 32.76 ± 0.23 | 28.0 ± 5.5 |
| 13 | 12 | 66.9 ± 13.5 | 60.8 ± 3.2 | 16.3 ± 3.1 | 16.5 ± 1.7 |
* Данные [43].
ЗАКЛЮЧЕНИЕ
В настоящей работе получены новые координационные соединения рутения с двумя биологически активными органическими фрагментами (полные аналоги противоопухолевого соединения NAMI-A), изучены их редокс-свойства и определены значения in vitro цитотоксичности соединений на клеточных линиях рака человека. Показано, что соединение 8, в состав которого входит фрагмент бексаротена, перспективно для химиотерапии аденокарциномы легкого, а соединение 13, содержащее лонидамин, – для химиотерапии аденокарциномы толстой кишки и аденокарциномы молочной железы человека.
Список литературы
Siddik Z.H. // Oncogene. 2003. V. 22. № 47. P. 7265. https://doi.org/10.1038/sj.onc.1206933
Wang D., Lippard S.J. // Nat. Rev. Drug Discov. 2005. V. 4. № 4. P. 307. https://doi.org/10.1038/nrd1691
Whiteside M.A., Piyathilake C.J., Bushell T.M. et al. // Nutrition and Cancer. 2006. V. 54. № 2. P. 274. https://doi.org/10.1207/s15327914nc5402_14
Abu-Surrah A S., Kettunen M. // Curr. Med. Chem. 2006. V. 13. № 11. P. 1337. https://doi.org/10.2174/092986706776872970
Hannon M.J. // Pure Appl. Chem. 2007. V. 79. № 12. P. 2243. https://doi.org/10.1351/pac200779122243
Florea A.-M., Büsselberg D. // Cancers. 2011. V. 3. № 1. P. 1351. https://doi.org/10.3390/cancers3011351
Wilson J.J., Lippard S.J. // Chem. Rev. 2014. V. 114. № 8. P. 4470. https://doi.org/10.1021/cr4004314
Giacchetti S., Perpoint B., Zidani R. et al. // J. Clin. Oncol. 2000. V. 18. № 1. P. 136. https://doi.org/10.1200/jco.2000.18.1.136
Galanski M., Jakupec M.A., Keppler B.K. // Curr. Med. Chem. 2005. V. 12. № 18. P. 2075. https://doi.org/10.2174/0929867054637626
Shpakovsky D.B., Shtil A.A., Kharitonashvili E.V. et al. // Metallomics. 2018. V. 10. № 3. P. 406. https://doi.org/10.1039/C7MT00286F
Simpson P.V., Desai N.M., Casari I. et al. // Future Med. Chem. 2019. V. 11. № 2. P. 119. https://doi.org/10.4155/fmc-2018-0248
Gonchar M.R., Matnurov E.M., Burdina T.A. et al. // J. Organomet. Chem. 2020. V. 919. P. 121 312. https://doi.org/10.1016/j.jorganchem.2020.121312
Milaeva E.R., Tyurin V.Y. // Pure Appl. Chem. 2017. V. 89. № 8. P. 1065. https://doi.org/10.1515/pac-2016-1130
Jakupec M.A., Arion V.B., Kapitza S. et al. // Int. J. Clin. Pharmacol. Ther. 2005. V. 43. № 12. P. 595. https://doi.org/10.5414/cpp43595
Rademaker-Lakhai J.M., van den Bongard D., Pluim D. et al. // Clin. Cancer Res. 2004. V. 10. № 11. P. 3717. https://doi.org/10.1158/1078-0432.Ccr-03-0746
Lentz F., Drescher A., Lindauer A. et al. // Anti-Cancer Drugs. 2009. V. 20. № 2. P. 97. https://doi.org/10.1097/CAD.0b013e328322fbc5
Henke M.M., Richly H., Drescher A. et al. // Int. J. Clin. Pharmacol. Ther. 2009. V. 47. № 1. P. 58. https://doi.org/10.5414/cpp47058
Vadori M., Pacor S., Vita F. et al. // J. Inorg. Biochem. 2013. V. 118. P. 21. https://doi.org/10.1016/j.jinorgbio.2012.09.018
Bergamo A., Gagliardi R., Scarcia V. et al. // J. Pharmacol. Exp. Ther. 1999. V. 289. № 1. P. 559.
Hartinger C.G., Jakupec M.A., Zorbas-Seifried S. et al. // Chem. Biodiversity. 2008. V. 5. № 10. P. 2140. https://doi.org/10.1002/cbdv.200890195
Pieper T., Borsky K., Keppler B.K. // Metallopharmaceuticals I: DNA Interactions / Eds. Clarke M.J., Sadler P.J. Berlin, Heidelberg: Springer, 1999. 199 p. https://doi.org/10.1007/978-3-662-03815-4_7
Nazarov A.A., Hartinger C.G., Dyson P.J. // J. Organomet. Chem. 2014. V. 751. P. 251. https://doi.org/10.1016/j.jorganchem.2013.09.016
Sava G., Gagliardi R., Bergamo A. et al. // Anticancer Res. 1999. V. 19. № 2a. P. 969.
Zorzet S., Bergamo A., Cocchietto M. et al. // J. Pharmacol. Exp. Ther. 2000. V. 295. № 3. P. 927.
Vacca A., Bruno M., Boccarelli A. et al. // Br. J. Cancer 2002. V. 86. № 6. P. 993. https://doi.org/10.1038/sj.bjc.6600176
Pluim D., van Waardenburg R.C.A.M., Beijnen J.H. et al. // Cancer Chemother. Pharmacol. 2004. V. 54. № 1. P. 71. https://doi.org/10.1007/s00280-004-0773-6
Kapitza S., Pongratz M., Jakupec M.A. et al. // J. Cancer Res. Clin. Oncol. 2005. V. 131. № 2. P. 101. https://doi.org/10.1007/s00432-004-0617-0
Galanski M., Arion V.B., Jakupec M.A. et al. // Curr. Pharm. Des. 2003. V. 9. № 25. P. 2078. https://doi.org/10.2174/1381612033454180
Kapitza S., Jakupec M.A., Uhl M. et al. // Cancer Letters. 2005. V. 226. № 2. P. 115. https://doi.org/10.1016/j.canlet.2005.01.002
Heffeter P., Pongratz M., Steiner E. et al. // J. Pharmacol. Exp. Ther. 2005. V. 312. № 1. P. 281. https://doi.org/10.1124/jpet.104.073395
Barnes K.R., Kutikov A., Lippard S. J. // Chem. Biol. 2004. V. 11. № 4. P. 557. https://doi.org/10.1016/j.chembiol.2004.03.024
Ang W.H., Khalaila I., Allardyce C.S. et al. // J. Am. Chem. Soc. 2005. V. 127. № 5. P. 1382. https://doi.org/10.1021/ja0432618
Dhar S., Liu Z., Thomale J. et al. // J. Am. Chem. Soc. 2008. V. 130. № 34. P. 11467. https://doi.org/10.1021/ja803036e
Mukhopadhyay S., Barnés C.M., Haskel A. et al. // Bioconjug. Chem. 2008. V. 19. № 1. P. 39. https://doi.org/10.1021/bc070031k
Pathak R.K., Marrache S., Choi J.H. et al. // Angew. Chem., Int. Ed. 2014. V. 53. № 7. P. 1963. https://doi.org/10.1002/anie.201308899
Neumann W., Crews B.C., Sárosi M.B. et al. // ChemMedChem. 2015. V. 10. № 1. P. 183. https://doi.org/10.1002/cmdc.201402353
Awuah S.G., Zheng Y.-R., Bruno P.M. et al. // J. Am. Chem. Soc. 2015. V. 137. № 47. P. 14854. https://doi.org/10.1021/jacs.5b10182
Suntharalingam K., Song Y., Lippard S.J. // Chem. Commun. 2014. V. 50. № 19. P. 2465. https://doi.org/10.1039/C3CC48740G
Agonigi G., Riedel T., Zacchini S. et al. // Inorg. Chem. 2015. V. 54. № 13. P. 6504. https://doi.org/10.1021/acs.inorgchem.5b00802
Raveendran R., Braude J.P., Wexselblatt E. et al. // Chem. Sci. 2016. V. 7. № 3. P. 2381. https://doi.org/10.1039/C5SC04205D
Nosova Y.N., Foteeva L.S., Zenin I.V. et al. // Eur. J. Inorg. Chem. 2017. V. 2017. № 12. P. 1785. https://doi.org/10.1002/ejic.201600857
Kenny R.G., Marmion C. // Chem. Rev. 2019. V. 119. № 2. P. 1058. https://doi.org/10.1021/acs.chemrev.8b00271
Okulova Y.N., Zenin I.V., Shutkov I.A. et al. // Inorg. Chim. Acta. 2019. V. 495. P. 119010. https://doi.org/10.1016/j.ica.2019.119010
Ang W.H., De Luca A., Chapuis-Bernasconi C. et al. // ChemMedChem. 2007. V. 2. № 12. P. 1799. https://doi.org/10.1002/cmdc.200700209
Ang W.H., Parker L.J., De Luca A. et al. // Angew. Chem., Int. Ed. 2009. V. 48. № 21. P. 3854. https://doi.org/10.1002/anie.200900185
Nazarov A.A., Gardini D., Baquié M. et al. // Dalton Trans. 2013. V. 42. № 7. P. 2347. https://doi.org/10.1039/C2DT31936E
Ji L., Zheng W., Lin Y. et al. // Eur. J. Med. Chem. 2014. V. 77. P. 110. https://doi.org/10.1016/j.ejmech.2014.02.062
Zheng W., Zhao Y., Luo Q. et al. // Sci. China: Chem. 2016. V. 59. № 10. P. 1240. https://doi.org/10.1007/s11426-016-0178-7
Nosova Y.N., Karlov D.S., Pisarev S.A. et al. // J. Organomet. Chem. 2017. V. 839. P. 91. https://doi.org/10.1016/j.jorganchem.2017.03.031
Носова Ю.Н. Дис. … канд. хим. наук. М., 2017. 171 с.
Reisner E., Arion V.B., Guedes da Silva M.F.C. et al. // Inorg. Chem. 2004. V. 43. № 22. P. 7083. https://doi.org/10.1021/ic049479c
Schluga P., Hartinger C.G., Egger A. et al. // Dalton Trans. 2006. № 14. P. 1796. https://doi.org/10.1039/B511792E
Reisner E., Arion V.B., Keppler B.K. et al. // Inorg. Chim. Acta. 2008. V. 361. № 6. P. 1569. https://doi.org/10.1016/j.ica.2006.12.005
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии