

Журнал неорганической химии, 2021, T. 66, № 3, стр. 317-324
Органоминеральные композиты гидроксиапатит кальция/альгинат калия: синтез, свойства
Н. А. Захаров a, *, Е. М. Коваль a, А. Д. Алиев b, Е. В. Шелехов c, М. Р. Киселёв b, В. В. Матвеев b, М. А. Орлов a, Л. И. Дёмина a, b, Т. В. Захарова d, Н. Т. Кузнецов a
a Институт общей и неорганической химии им. Н.С. Курнакова РАН
119991 Москва, Ленинский пр-т, 31, Россия
b Институт физической химии и электрохимии им. А.Н. Фрумкина РАН
119071 Москва, Ленинский пр-т, 31, Россия
c Национальный исследовательский технологический университет “МИСИС”
119049 Москва, Ленинский пр-т, 4, Россия
d Российский университет транспорта “МИИТ”
127994 Москва, ул. Образцова, 9, Россия
* E-mail: zakharov@igic.ras.ru
Поступила в редакцию 21.08.2020
После доработки 14.09.2020
Принята к публикации 18.09.2020
Аннотация
В ходе совместного осаждения гидроксиапатита кальция Ca10(PO4)6(OH)2 (ГА) и природного биополимера альгината калия (АК) [С6Н7КО6]n из водного раствора в системе CaCl2–(NH4)2HPO4–NH3–[С6Н7КО6]n–H2O (25°С) синтезированы органоминеральные композиты ГА/АК на основе нанокристаллического ГА с содержанием 0.1, 0.2, 0.3, 0.4 мас. % АК. Продукты синтеза идентифицированы методами рентгенофазового и термического анализа, инфракрасной спектроскопии, сканирующей и просвечивающей электронной микроскопии, электронной спектроскопии для химического анализа.
ВВЕДЕНИЕ
Создание органоминеральных композитов (ОМК) на основе биосовместимых фосфатов кальция и биополимеров является актуальным направлением разработки перспективных материалов для костных имплантатов с улучшенными характеристиками [1]. Гидроксиапатит кальция Ca10(PO4)6(ОН)2 (ГА) – кристаллохимический аналог неорганической компоненты костной ткани млекопитающих [2]. Он обладает характеристиками биосовместимости и биоактивности. Костная ткань, являясь природным наноразмерным ОМК, включает в свой состав биополимеры (в основном коллаген), клетки и другие нативные ткани [3]. Моделирование состава природного ОМК – костной ткани – достигается с использованием биополимеров, позволяющих имитировать наиболее характерные свойства нативной костной ткани [4–6].
Одним из перспективных представителей подобных биополимеров является гетерополимер альгиновая кислота (С6Н8О6)n и ее соли [7–10]. Крупномасштабное производство альгиновой кислоты основано на извлечении ее из красных, бурых и некоторых зеленых водорослей. Широкое использование альгиновой кислоты и ее производных в промышленности обусловлено их способностью к набуханию, вязкостью их растворов и высокой способностью взаимодействать с различными структурами [11–13]. В настоящее время альгиновая кислота и соединения на ее основе используются в текстильной (50%) и пищевой (30%) промышленности, медицине, косметике, фармакологии (20%) [14].
Особенно перспективной для использования в фармакологии является соль альгиновой кислоты – альгинат калия [С6Н7KО6]n (АК). Он отличается рядом уникальных свойств [15, 16], таких как биосовместимость, биоактивность, иммуномодулирующее действие, способность к обволакивающему воздействию и др. [17, 18].
Перспективным подходом для решения задачи создания ОМК на основе ГА и биополимеров является совместное осаждение солей кальция, фосфора и биополимеров из растворов различного состава. Это позволяет в ходе решения задачи синтеза ОМК найти подходы к моделированию процессов биоминерализации в ходе остеогенеза. Такие методы направленного синтеза будут способствовать созданию новых материалов с регулируемыми в ходе синтеза и последующей обработки свойствами (размер и морфология кристаллов фосфатов кальция, их растворимость, пористость, биосовместимость и др.).
Ниже описан синтез из водных растворов в системах CaCl2–(NH4)2HPO4–NH3–[С6Н7КО6]n–H2O (25°С) ОМК ГА/АК, содержащих 0.1, 0.2, 0.3, 0.4 мас. % АК, и проанализированы с помощью методов физико-химического анализа взаимосвязи состав–условия синтеза–структура–дисперсность–свойства.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Материалы. В качестве исходных реактивов для синтеза ОМК ГА/АК использовали водные растворы CaCl2 (ч. д. а.), (NH4)2HPO4 (ч.), аммиака и дистиллированную воду.
Источником АК служил водный раствор калиевой соли альгиновой кислоты водоросли ламинарии производства АО “Архангельский опытный водорослевый комбинат” ТУ 10.89. 19-047-00462769-2016. Такой АК соответствует индексу европейской кодификации Е402 для пищевых добавок, допущенных к применению в пищевой промышленности Российской Федерации в качестве вспомогательного средства для производства пищевых продуктов (загустители, стабилизаторы, средства желеобразования), в фармакологии и косметологии. Использованный для синтеза АК являлся волокнистым, с мелкими гранулами и пластинами порошок без запаха бледно-желтого цвета. Содержание основного вещества в использованном АК было не менее 90%, плотность составляла 1.60 г/см3. Исходный АК был образован полисахаридными цепями мануроновой и гиалуроновой кислот и калием, эмпирическая формула [С6Н7KО6]n. В качестве исходного реактива использовали 1%-ный водный раствор АК.
Синтез объектов исследования. Процедура синтеза ОМК ГА/АК в системах CaCl2–(NH4)2HPO4–NH3–[С6Н7КО6]n–H2O (25°С), выбранные значения рН и соотношений CaCl2/(NH4)2HPO4 в исходных смесях соответствовали установленным ранее [19] оптимальным условиям образования ГА. В ходе изучения водных систем CaCl2–(NH4)2HPO4–NH3–[С6Н7КО6]n–H2O соблюдались условия, при которых содержание (NH4)2HPO4 во всех пробах составляло 0.05 моль/л; отношение компонентов в исходных смесях выдерживали равным n1 = CaCl2/(NH4)2HPO4 = = 1.67; значение рН поддерживали в пределах 10–10.5; концентрация [С6Н7КО6]n составляла 0.001–0.004 моль/л, что соответствует отношению n2 = [С6Н7КО6]n/(NH4)2HPO4 = 0.1–0.4. Добавление раствора АК в состав реагирующих смесей (200 мл) проводили в последнюю очередь, после чего следовало перемешивание реагирующих растворов в течение 14 сут магнитной мешалкой при комнатной температуре.
Химический анализ продуктов синтеза. По окончании реакции синтеза в равновесных жидких фазах проводили измерение рН (прибор рН121) и определение содержания ионов Ca2+ (комплексонометрический метод вытеснения в комбинации с комплексонатом цинка с эриохромом черным Т в качестве индикатора [20]) и ${\text{PO}}_{{\text{4}}}^{{{\text{3}}--}}$ (весовой хинолинмолибдатный метод) с целью установления соотношения n3 = Ca2+/${\text{PO}}_{{\text{4}}}^{{{\text{3}}--}}$ в образовавшихся твердых фазах. После декантации жидкой фазы образовавшийся осадок отфильтровывали, промывали дистиллированной водой до полного удаления ионов хлора и сушили на воздухе при комнатной температуре с целью получения образцов для физико-химического анализа.
Приборные методы физико-химического анализа
Рентгенофазовый анализ (РФА). Определение фазового состава, кристаллической структуры и морфологии нанокристаллов ГА (НКГА) порошкообразных образцов проводили с использованием автоматизированного рентгеновского дифрактометра ДРОН-4, сфокусированного по Бреггу–Брентано [21], с графитовым монохроматором на дифрагированном пучке, управляемым с помощью программы EXPRESS. Измерения проводили на CuKα-излучении в режиме пошагового сканирования с шагом 0.1°; время экспозиции на одну точку составляло 3 с.
Для качественного и количественного рентгенофазового анализа использовали программы PHAN и PHAN% (модифицированный полнопрофильный анализ, позволяющий оценить размер блоков и величину микродеформаций кристаллической решетки) с известным элементным составом образцов. Банк данных содержал более 110 тысяч карточек в формате картотеки JCPDS [22].
Инфракрасная (колебательная) спектроскопия (ИКС). ИК-спектры диффузного отражения продуктов синтеза регистрировали в диапазоне 4000–400 см–1 с шагом сканирования 1 см–1 с использованием ИК-фурье-спектрометра Nexus (фирмы Nicolet, США). Образцы для измерений были оформлены в виде спрессованных дисков смеси продуктов синтеза с KBr.
Термогравиметрический анализ (ТГА) образцов проводили с помощью термоанализатора NETZSCH Simultaneous Thermal Analyzer STA 409 на воздухе в интервале температур 20–1000°С. Скорость нагрева составляла 10 град/мин, масса навески 5–20 мг.
Сканирующая электронная микроскопия (СЭМ), электронная спектроскопия для химического анализа (ЭСХА). Морфологию поверхности образцов ОМК изучали с использованием микроскопа CamScanS4. Рентгеновский микроанализ (ЭСХА) изученных объектов был выполнен с помощью энергодисперсионного микроанализатора Link Analytical. Измерения проводили при токе пучка 10–10 А.
Просвечивающая электронная микроскопия (ПЭМ). Наблюдение наноструктуры образцов и НКГА в составе ОМК ГА/АК проводили методом ПЭМ с использованием электронного микроскопа JEOL JEM 1210.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Химический анализ. Результаты химического анализа методом остаточных концентраций и измерения рН свидетельствуют (табл. 1) о практически полном взаимодействии исходных прекурсоров на протяжении выбранного времени (14 сут) синтеза и незначительном (на уровне погрешности эксперимента) содержании ионов Са2+, ${\text{PO}}_{{\text{4}}}^{{{\text{3}}--}}$ в образующейся после отстаивания продукта жидкой фазе.
Таблица 1.
Остаточные концентрации, состав продуктов синтеза и рН в системе CaCl2–(NH4)2HPO4–NH3–[С6Н7КО6]n–H2O (25°С)
| № п/п |
Содержание АК, мас. % | Остаточные концентрации, моль/л | pH | Ca/P (расчет) | Состав продуктов синтеза | |
|---|---|---|---|---|---|---|
| Ca2+ | P${\text{O}}_{4}^{{3 - }}$ | |||||
| 1 | 0.1 | Cледы | 10.3 | 1.67 | Ca10(PO4)6(OH)2 · 0.1АК · 6.8Н2О | |
| 2 | 0.2 | » | 10.5 | 1.67 | Ca10(PO4)6(OH)2 · 0.2АК · 8.3Н2О | |
| 3 | 0.3 | » | 10.2 | 1.67 | Ca10(PO4)6(OH)2 · 0.3АК · 9.6Н2О | |
| 4 | 0.4 | » | 10.3 | 1.67 | Ca10(PO4)6(OH)2 · 0.4АК · 10.8Н2О | |
Рассчитанные отношения n4 = Са2+/${\text{PO}}_{{\text{4}}}^{{{\text{3}}--}}$ в твердых фазах при выбранных условиях синтеза (n1 = 1.67, рН 10–10.2, время перемешивания 14 сут) во всех проанализированных случаях составляли 1.67, что соответствует образованию фосфатов кальция со структурой ГА и стехиометрическим отношением Са/Р. На основании результатов химического анализа можно сделать вывод об образовании в ходе синтеза ОМК ГА/АК состава Ca10(PO4)6 · [С6Н7КО6]х(OH)2 · zH2O, где х = 0.1–0.4; z = 6.8–10.8.
ИК-спектры отражения. ИК-спектры продуктов синтеза (рис. 1 ) характеризуются типичными для ГА полосами валентных (1093, 1038 и 963 см–1) и деформационных (603, 567 см–1) колебаний группировок ${\text{PO}}_{{\text{4}}}^{{{\text{3}}--}}$ в составе ГА. Деформационные колебания ${\text{PO}}_{{\text{4}}}^{{{\text{3}}--}}$ проявляются в колебательном спектре поглощения при 604, 566 и 470 см–1. Полоса ν(ОН) при 3570 см–1, как и в спектрах нативных апатитов [2], характеризуется незначительной интенсивностью, относительная величина которой возрастает после термической обработки синтезированных образцов.
Рис. 1.
ИК-спектры отражения: а – альгината калия ([С6Н7КО6]n); б – композита состава Са1(PO4)6(ОН)2 · · [С6Н7КО6]х(OH)2 · zH2O, х = 0.3, z = 6.8–10.8; в – композита, соответствующего случаю б, подвергнутого термическому воздействию (1000°С, 1 ч).
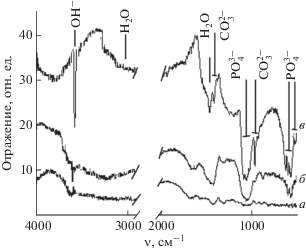
Значительный фон в области 3500–2900 см–1 и полоса деформационных колебаний Н–О–Н при ~1671 см–1 (рис. 1 ) обусловлены высокой адсорбционной способностью развитой поверхности НКГА в составе ОМК. Полосы поглощения карбоната в составе синтезированных ОМК фиксировали приблизительно при 1500, 1458, 1420 и 875 см–1. Сравнение полос поглощения для ОМК, полученных в настоящей работе, с литературными данными для иных типов апатитов [2] позволяет сделать вывод о преимущественном замещении ОН–-групп апатита ионами карбоната и свидетельствует об образовании в ходе синтеза НКГА преимущественно с А-типом замещения (замещение ОН-групп). Ввиду незначительного содержания АК в составе ОМК ГА/АК его спектральные характеристики не сказывались существенным образом на колебательных спектрах ОМК. Это, как правило, характерно и для ОМК на основе ГА и биополимеров иного состава при сопоставимом содержании последнего в составе ОМК.
Рентгенофазовый анализ. Синтезированные в составе ОМК ГА/АК апатиты, по данным РФА (табл. 2), характеризовались как однородные твердые фазы на основе ГА (пр. гр. P63/m). Условия синтеза обеспечивали отсутствие в продуктах синтеза посторонних фаз (СаСО3, СаО, Са3(РО4)3), свидетельствуя о полном прохождении реакции. Уширение дифракционных линий (рис. 2 ) свидетельствовало о нанокристаллическом состоянии ГА в составе ОМК ГА/АК. Термическая обработка также не вызывала образования посторонних фаз, увеличивая в то же время степень кристалличности ГА и улучшая разрешение его дифракционных линий (рис. 2 ).
Таблица 2.
Кристаллографические и морфологические характеристики ГА в составе ОМК ГА/АК
| № п/п | Параметры ячейки, Å | Размер блока Коши, нм (1) | Брутто-формула | ||
|---|---|---|---|---|---|
| а | с | ||с | $ \bot $с | ||
| 1 | 9.4236 | 6.8786 | 6.87 | 8.2 | Ca10(PO4)6 · 0.1[С6Н7КО6] · (OH)2 · 6.8H2O |
| 2 | 9.4247 | 6.8794 | 11.5 | 7.4 | Ca10(PO4)6 · 0.2[С6Н7КО6] · (OH)2 · 8.3H2O |
| 3 | 9.4242 | 6.8782 | 15.4 | 8.0 | Ca10(PO4)6 · 0.3[С6Н7КО6] · (OH)2 · 9.6H2O |
| 4 | 9.4264 | 6.8747 | 12.2 | 7.2 | Ca10(PO4)6 · 0.4[С6Н7КО6] · (OH)2 · 10.8H2O |
| 5 | 9.48 | 6.884 | – | – | Ca10(PO4)6(OH)2 (2) |
(1) Размер блока Коши параллельно (||с) и перпендикулярно ($ \bot $с) гексагональной оси с. (2) Параметры элементарной ячейки ГА в соответствии с JCPDS (№ 9–432) [22].
Рис. 2.
Дифрактограммы: a – Са10(PO4)6(ОН)2 · · [С6Н7КО6]х(OH)2 · zH2O, где х = 0.3; z = 9.6; б – того же образца, подвергнутого термической обработке (1000°С, 1 ч).
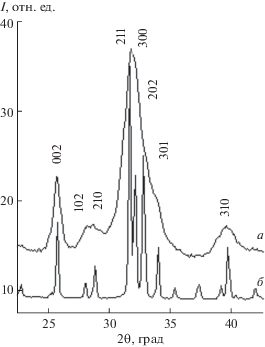
Параметры элементарных ячеек ГА в составе ОМК ГА/АК находятся в удовлетворительном соответствии с данными JCPDS (№ 9-432) [22]. НКГА удлинены вдоль гексагональной оси с и имеют размеры и кристаллографические характеристики, близкие к таковым для НКГА нативной кости [23] (табл. 2).
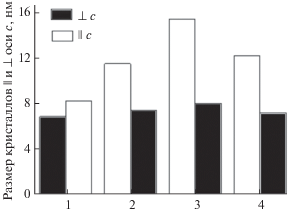
Увеличение содержания АК в составе ОМК ГА/АК даже в незначительных количествах (0.1–0.4 мас. %) сопровождалось ощутимым удлинением НКГА вдоль гексагональной оси c (рис. 3 ). При этом их толщина в направлении, перпендикулярном оси c, изменялась незначительным образом. Рост содержания АК сопровождался небольшим уменьшением параметра c и незначительным ростом параметра a элементарной ячейки НКГА (табл. 2).
Рис. 3.
Морфологические характеристики НКГА в составе ОМК ГА/АК с содержанием 0.1 (1), 0.2 (2), 0.3 (3) и 0.4 (4) мас. % АК.
Термогравиметрический анализ образцов ОМК ГА/АК. Кривые термического разложения исходного АК (рис. 4 а) характеризуются на начальном этапе потерей веса в области 100°С (~14.5%), связанной с выделением адсорбированной воды (эндотермический эффект 227.7 Дж/г). В области более высоких температур (~260, 350°С) происходит выгорание органической компоненты с потерей веса (от начального его значения) при указанных температурах соответственно ~50 и 63% и интенсивным экзотермическим эффектом (~947 Дж/г) при 342°С. Нагревание выше 600°С сопровождается характерным для органических биополимеров процессом декарбонизиции [24], дальнейшей потерей веса (~75%) и значительным экзотермическим эффектом (1673 Дж/г).
Перечисленные особенности термических характеристик АК отражаются и на термическом поведении ОМК ГА/АК (рис. 4 б; образец состава Ca10(PO4)6(OH)2 · 0.2АК · 8.3Н2О). Незначительное содержание (0.2 мас. %) АК в образцах этого ОМК накладывает отпечаток на его термические характеристики. Характерные для индивидуального АК эффекты выделения адсорбированной воды (~100°С), выгорания органической компоненты (~260, 350°С) и декарбонизации (выше 600°С) явно не выражены. Термические эффекты агломерации НКГА при повышенных температурах незначительны, что не позволяет выявить их на фоне других термических эффектов.
Рис. 4.
Зависимости ТГА и ДСК АК (С6Н8О6)n (a) и образца ОМК ГА/АК состава Ca10(PO4)6(OH)2 · 0.2АК · 8.3Н2О (б).
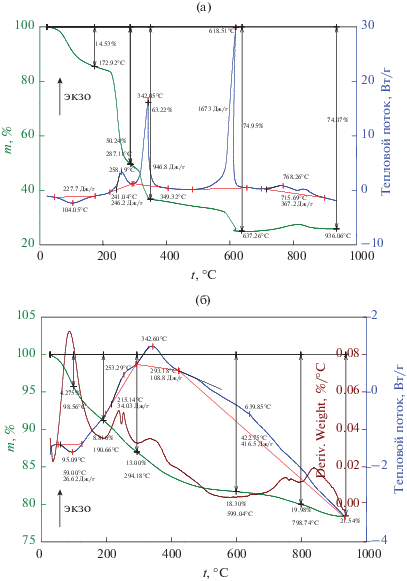
Результаты термического анализа позволяют сделать заключение о практическом отсутствии изменения характеристик АК в составе ОМК ГА/АК до температуры 200°С. По данным термического анализа, предельные температуры, при которых ОМК способны сохранять свои стабильные характеристики, не превышают 250°С.
Электронная микроскопия (СЭМ, ПЭМ) и ЭСХА продуктов синтеза. Результаты ЭСХА (рис. 5 ) свидетельствуют об однородности состава по всему объему синтезированных ОМК ГА/АК и находятся в удовлетворительном соответствии с данными химического анализа (табл. 1) по основным элементам ОМК (кальций, фосфор, кислород, углерод). Продукты синтеза после высыхания на воздухе представляют собой твердые черепки. Помол продуктов синтеза (ступка, шаровая мельница) приводит к образованию частиц с размером от единиц до десятков микрон (рис. 5 ), сохраняющих свою форму и не отличающихся друг от друга по химическому составу. Изменение состава ОМК ГА/АК не влияет на характер измельчения продуктов синтеза. Влияние увеличенной (за счет присутствия АК) вязкости раствора сказывается в определенной мере на образовании пористой структуры твердых образцов после высыхания ОМК (рис. 5 в, 5г).
Рис. 5.
Результат ЭСХА (а) и картины СЭМ (б, в, г) в прямом (б, в) и обратном (г) свете образца ОМК ГА/АК состава Ca10(PO4)6(OH)2 · 0.2АК · 8.3Н2О.
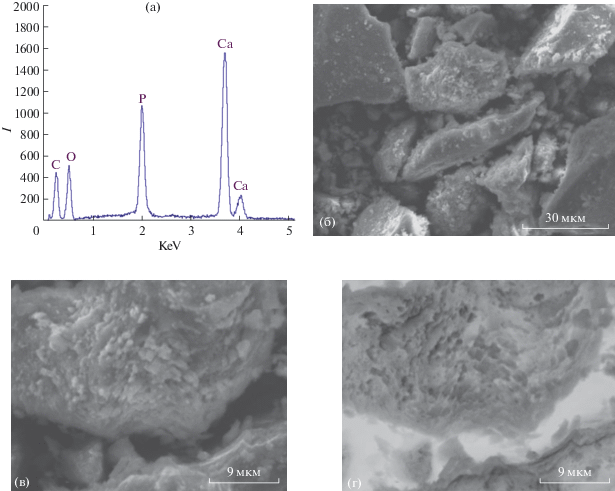
По-видимому, повышенная вязкость исходного раствора для синтеза сказывается и на росте НКГА, вызывая склонность к возникновению аморфной фазы образующегося апатита. О возможности подобного явления указывалось ранее [25] при рассмотрении влияния различных факторов на образование аморфного ГА в ходе его синтеза из водных растворов. В случае образования ОМК ГА/АК в описанной в настоящей работе системе результаты ПЭМ явно указывают (рис. 6 ) на присутствие аморфной составляющей в образующихся продуктах синтеза. Об этом свидетельствуют как неразвитые формы НКГА, так и нерельефная форма агломератов НКГА на картинах ПЭМ, отличающие их от НКГА композитов, включающих в свой состав менее вязкие биополимеры (фиброин шелка [26], метилцеллюлозу [27]) либо наноуглеродные материалы (многостенные углеродные нанотрубки [28], оксид графена [29]). По-видимому, вязкость растворов для синтеза сказывалась и на морфологии НКГА в ОМК ГА/АК. В отличие от упомянутых композитов на основе ГА [25–28], НКГА в ОМК ГА/АК не были вытянуты вдоль гексагональной оси с, а были склонны к росту в перпендикулярном направлении. Следует отметить, что присутствие доли метастабильной аморфной фракции в образующихся продуктах синтеза будет способствовать желательному увеличению растворимости апатита в составе ОМК ГА/АК, увеличивая степень перспективности его практического использования.

ЗАКЛЮЧЕНИЕ
В ходе совместного осаждения солей кальция, фосфора и биополимера альгината калия (АК) [С6Н7КО6]n определено влияние АК на осаждение из водных растворов состава CaCl2–(NH4)2HPO4–NH3–[С6Н7КО6]n–H2O (25°С) гидроксиапатита кальция (Ca10(PO4)6(ОН)2).
Синтезированы органоминеральные нанокомпозиты на основе ГА и АК с содержанием 0.1, 0.2, 0.3, 0.4 мас. % АК, продукты синтеза идентифицированы методами химического, рентгенофазового и термогравиметрического анализа, ИК-спектроскопии, сканирующей и просвечивающей электронной микроскопии и электронной спектроскопии для химического анализа.
На основе анализа физико-химических характеристик установлены взаимосвязи состав–условия синтеза–структура–дисперсность–свойства, позволяющие осуществить подходы для направленного синтеза наноразмерных ОМК ГА/АК, моделирующих состав и свойства нативной костной ткани.
Высказано предположение, что затруднение развития НКГА в направлении гексагональной оси с и возникновение аморфной фракции ГА на картинах ПЭМ может быть вызвано повышенной вязкостью водного раствора АК для синтеза ОМК.
Рассмотрены возможности применения синтезированных ОМК в качестве материалов для костных имплантатов с улучшенными характеристиками.
Список литературы
Dorozhkin S.V. // BIO. 2011. V. 1. P. 1.
Elliot J.C. Structure, chemistry of apatites and other calcium orthophosphates. Amsterdam: Elsevier Science, 1994. 350 p.
Касавина Б.С., Торбенко В.П. Жизнь костной ткани. М.: Наука, 1979. 176 с.
Sancilio S., Gallorini M., Di Nisio C. et al. // Stem Cells Intern. 2018. V. 1. P. 1.
Kobayashi M., Sakane M., Abe T. et al. // Bioceram. Develop. Applic. 2012. V. 2. P. 1.
Kamalaldina N.A., Yahya B.H., Nurazreenab A. // Proc. Chem. 2016. V. 19. P. 297.
Титов А.М. Целительные свойства морских водорослей: повышение иммунитета, нормализация обмена веществ, защита от рака. СПб.: Нева, 2004. 125 с.
Suarez-Gonzalez D., Barnhart K., Saito E. et al. // J. Biomed. Mater. Res. A. 2010. V. 95A. № 1. P. 221.
Rajesh R., Ravichandran Y.D. et al. // Int. J. Nanomedicine. 2015. V. 10. P. 7.
Cardoso D.A., Ulset A.-S., Bender J. et al. // Macrom. Biosc. 2014. V. 14. № 6. P. 872.
Сарафанова Л.А. Пищевые добавки: энциклопедия. СПб.: ГИОРД, 2004. 809 с.
Onoyima C.C., Okibe F.G., Anweting I.B. et al. // Trends Sc. Technol. J. 2017. V. 2. № 1A. P. 261.
Obara S., Yamauchi T., Tsubokawa N. // Polym. J. 2010. V. 42. P. 161.
Криштанова Н.А., Сафонова М.Ю., Болотова В.Ц. и др. // Вестник ВГУ. Серия: Химия. Биология. Фармация. Санкт-Петербургская гос. хим.-фармац. акад. 2005. № 1. С. 212.
Ботанико-фармакологический словарь. М.: Высшая школа, 1990. 314 с.
Manatunga D.C., de Silva Rohini M., de Silva K.M.N. et al. // Chem. Centr. J. 2018. V. 12. № 119. P. 1.
Sellimi S., Younes I., Ayed H.B. et al. // Int. J. Biol. Macromol. 2015. V. 72. P. 1358.
Wang Y.-P., Liao Y.-T., Liu C.-H. et al. // Biointerphases. 2015. V. 10. № 2. P. 1.
Чумаевский Н.А., Орловский В.П., Ежова Ж.А. и др. // Журн. неорган. химии. 1992. Т. 37. № 7. С. 881.
Шварценбах Г., Флашка Г. Комплексонометрическое титрование. М.: Химия, 1970. С. 172.
Горелик С.С., Скаков Ю.А., Расторгуев Л.Н. Рентгенографический и электронно-оптический анализ. М.: МИСиС, 2002. 360 с.
Powder diffraction file (inorganic phases). Joint Committee on Powder Diffraction Standards (JCPDS) File № 9-432, International Centre of Diffraction Data, Newton Square, PA, 1980.
Hench L.L. // J. Am. Ceram. Soc. 1991. V. 74. № 7. P. 1487.
Zakharov N.A., Ezhova Zh.A., Koval E.M. et al. // Russ. J. Inorg. Chem. 2015. V. 60. № 12. P. 1460.
Rey C., Combes C., Drouet C. et al. // Mater. Sci. Eng. 2007. V. 27. P. 198.
Zakharov N.A., Demina L.I., Aliev A.D. et al. // Inorg. Mater. 2017. V. 53. № 3. P. 333.
Zakharov N.A., Sentsov M.Yu., Kiselev M.R. et al. // Protect. Met. Phys. Chem. Surf. 2016. V. 52. № 1. P. 89.
Zakharov N.A., Sentsov M.Yu., Chalykh A.E. et al. // Protect. Met. Phys. Chem. Surf. 2013. V. 49. № 1. P. 80.
Zakharov N.A., Tkachev A.G., Demina L.I. et al. // Protect. Met. Phys. Chem. Surf. 2016. V. 52. № 4. P. 665.
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии