

Журнал неорганической химии, 2021, T. 66, № 2, стр. 134-140
Синтез нитрилиевых производных клозо-дека- и додекаборатного анионов [BnHn–1NCR]– (n = 10, 12) микроволновым методом
А. В. Нелюбин a, И. Н. Клюкин a, А. П. Жданов a, *, М. С. Григорьев b, К. Ю. Жижин a, Н. Т. Кузнецов a
a Институт общей и неорганической химии им. Н.С. Курнакова РАН
119991 Москва, Ленинский пр-т, 31, Россия
b Институт физической химии и электрохимии им. А.Н. Фрумкина РАН
119991 Москва, Ленинский пр-т, 31, Россия
* E-mail: zhdanov@igic.ras.ru
Поступила в редакцию 26.06.2020
После доработки 10.08.2020
Принята к публикации 15.08.2020
Аннотация
Предложены методы получения нитрилиевых производных высших кластерных анионов бора с помощью методологии СВЧ-синтеза. Предложенные подходы использованы для синтеза производных на основе высококипящих и твердых органических нитрилов. Впервые получены производные аниона [B10H10]2– на основе динитрилов и выделены в индивидуальном виде производные [B12H11NCCH3]– и [B12H11NCC2H5]–. Полученные соединения исследованы методами мультиядерной спектроскопии ЯМР, ИК-спектроскопии поглощения, элементного анализа.
ВВЕДЕНИЕ
Разработка новых и усовершенствование уже существующих синтетических методов – ключевая задача экспериментальной неорганической химии [1]. В настоящее время разработано множество подходов к функционализации кластерных соединений бора и их аналогов: карборанов, металлоборанов и гетероборанов [2]. Данные методы лежат на стыке самых современных разработок в области неорганической и элементоорганической химии. Основной целью экспериментальных исследований является создание универсальных и высокоселективных синтетических протоколов, позволяющих получать кластерные соединения с заданными свойствами. Интерес к химии борсодержащих кластеров продиктован возможностью применения данных соединений в медицине, катализе, создании материалов для нелинейной оптики [3, 4].
Химия кластерных соединений бора включает в себя множество разнообразных синтетических методов благодаря разнообразию строения борных полиэдров, а также возможности включения в молекулярный остов гетероатомов. Так, для карборанов характерны подходы к функционализации, основанные на гетеро- или гомологическом разрыве связи C–H [5]. Для металлоборанов разработаны методы, связанные с изменением лигандного окружения центрального атома металла [6, 7]. Для химии кластерных анионов бора общего вида [BnHn]2–, где n = 6–12, наиболее характерны процессы, происходящие с разрывом экзо-полиэдрических связей B–H [8]. Эти процессы могут реализовываться по механизму электрофильного, радикального или электрофильно-индуцируемого нуклеофильного замещения (EINS) [9]. Последний класс процессов получил наибольшее распространение благодаря возможности введения широкого круга экзополиэдрических заместителей, высокой регио- и стереоселективности, а также простоте аппаратного оформления.
В настоящее время разработаны подходы к получению клозо-боратных анионов с экзополиэдрическими связями B–O, B–S, B–N [10, 11]. Особый интерес представляют нитрилиевые производные на основе клозо-декаборатного аниона общего вида [B10H9NCR]–. Данный класс соединений содержит в своем составе активированную кратную связь С–N, способную вступать в реакции нуклеофильного присоединения [12–14]. Процесс получения нитрилиевых производных клозо-боратного аниона основан на взаимодействии клозо-декаборатного аниона [B10H10]2– с органическими нитрилами в присутствии трифторуксусной кислоты CF3COOH [15–17] в качестве электрофильного индуктора. Альтернативный подход основан на использовании аниона [B10H11]– в качестве стартового борсодержащего синтона [18]. Данный метод позволяет проводить реакцию без дополнительного электрофильного индуктора.
Микроволновое излучение (нагревание) успешно применяется для синтеза неорганических, органических и координационных соединений и получения материалов на их основе [19–24]. В химии борных кластеров применение СВЧ-синтеза на данный момент не так широко распространено и чаще применяется для модификации карборанов [25, 26], металлоборанов [27] и додекаборатных анионов с исчерпывающей степенью замещения [28–30]. Известно несколько примеров использования методологии микроволнового синтеза для направленной модификации карборанов [31] и кластерных анионов бора [32, 33].
Настоящая работа посвящена направленному синтезу нитрилиевых производных клозо-дека- и додекаборатного анионов с использованием СВЧ-нагревания.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Элементный анализ на углерод, водород и азот осуществляли на автоматическом газовом анализаторе CHNS-3 FA 1108 Elemental Analyser (Carlo Erba). Определение бора и гафния методом ICP MS выполнено на атомно-эмиссионном спектрометре с индуктивно-связанной плазмой iCAP 6300 Duo в ЦКП “Научно-аналитического центра ФГУП “ИРЕА” Национального исследовательского центра Курчатовский институт”.
ИК-спектры соединений записывали на ИК-Фурье-спектрофотометре Инфралюм ФТ08 (НПФ АП “Люмекс”) в области 4000–400 см–1 с разрешением 1 см–1. Образцы готовили в виде раствора в хлороформе.
Спектры ЯМР 1H, 11B, 13C растворов исследуемых веществ в CD3CN записывали на импульсном Фурье-спектрометре Bruker MSL-300 (Германия) на частотах 300.3, 96.32 и 75.49 МГц соответственно с внутренней стабилизацией по дейтерию. В качестве внешних стандартов использовали тетраметилсилан или эфират трехфтористого бора.
Рентгеноструктурный анализ (NBu4)[2-B10H9NCnC3H7] выполнен в ЦКП ИФХЭ РАН на автоматическом четырехкружном дифрактометре с двумерным детектором Bruker KAPPA APEX II (излучение MoKα) [34] с использованием фрагмента кристалла размерами 0.30 × 0.20 × 0.08 мм при температуре 100 K.
Параметры элементарной ячейки уточнены по всему массиву данных [35]. Структура расшифрована прямым методом [36] и уточнена полноматричным методом наименьших квадратов [37] по F2 по всем данным в анизотропном приближении для всех неводородных атомов (кроме разупорядоченных, если такие имеются). Атомы H кластера бора локализованы из разностного Фурье-синтеза электронной плотности и уточнены изотропно без каких-либо ограничений. Атомы H групп CH, CH2 и CH3 размещены в геометрически вычисленных позициях и уточнены с изотропными температурными параметрами, равными 1.2Uэкв атома C для CH, CH2 и 1.5Uэкв атома C для CH3. Координаты атомов депонированы в Кембриджской кристаллографической базе данных (CCDC 2 012 227).
Растворители и реагенты марки “х. ч.” и “ос. ч.” использовали без дополнительной очистки.
Синтез нитрилиевых производных аниона [B10H10]2–. Растворяли 0.361г (1.0 ммоль) (NBu4)[B10H11] в 50 мл CH2Cl2. Полученный раствор помещали в реактор и добавляли 1.0 мл в случае пропионитрила и бутиронитрила или 1.1 ммоль в случае сукцино-, глутаро-, адипонитрила, 1-цианоадамантана. Реактор продували сухим аргоном и герметизировали. Реакционную смесь нагревали в течение 1.5 ч под давлением при 50°С и мощности микроволнового излучения 100 Вт. После завершения процесса синтеза реакционную смесь охлаждали до комнатной температуры и упаривали на роторном испарителе. К полученному продукту добавляли 20 мл петролейного эфира и обрабатывали ультразвуком в течение 10 мин. Целевой продукт отделяли декантацией и сушили в эксикаторе над P2O5.
Синтез нитрилиевых производных аниона [B12H12]2–. Растворяли 0.626 г (1.0 ммоль) (NBu4)2[B12H12] в 50 мл соответствующего нитрила. Полученный раствор помещали в реактор и добавляли 0.19 мл (2.5 ммоль) CF3COOH. Реактор продували сухим аргоном и герметизировали. Реакционную смесь нагревали в течение 2 ч под давлением при 80°С и мощности микроволнового излучения 100 Вт. После завершения процесса синтеза реакционную смесь охлаждали до комнатной температуры, концентрировали на роторном испарителе до сиропообразного состояния и добавляли 20 мл ледяной уксусной кислоты. Полученный продукт отфильтровывали, промывали на фильтре холодным диэтиловым эфиром и сушили в эксикаторе над P2O5.
(NBu4)[2-B10H9NC(CH2)2CN] (1). ИК-спектр (CHCl3, см–1): 2489 ν(B–H), 2359 ν(C≡N (B–N)), 2260 ν(C≡N), 1032 δ(B–B–H); 11B ЯМР (CD3CN, δ, м.д.): –0.1 (д, 1B, B(10), JB–H = 149 Гц), –2.9 (д, 1B, B(1), JB–H = 150 Гц), –22.6 (с, 1B, B(2)), ‒26.6 (д, 3B, B(4,7,8), JB–H = 108 Гц), –29.1 (д, 4B, B(3,5,6,9), JB–H = 134 Гц); 1Н ЯМР (CD3CN, δ, м.д.): –1.01–1.55 (м, 9Н, В10Н9), 3.22 (т, 2H, B–NC–CH2–CH2–CN, J = 7 Гц), 3.15 (м, 8H, NBu4), 2.77 (т, 2H, B–NC–CH2–CH2–CN, J = 6 Гц), 1.60 (м, 8H, NBu4), 1.35 (м, 8H, NBu4), 0.95 (м, 12H, NBu4); 13С ЯМР (CD3CN, δ, м.д.): 126.2 (B–NC–CH2–CH2–CN), 119.2 (B–NC–CH2–CH2–CN), 59.4 (NBu4), 35.6 (B–NC–CH2–CH2–CN), 27.4 (B–NC–CH2–CH2–CN), 24.5 (NBu4), 20.4 (NBu4), 14.0 (NBu4).
| C | H | N | B | |
|---|---|---|---|---|
| Найдено, %: | 54.91; | 11.21; | 9.61; | 23.9. |
| Для C20H49B10N3 (М = 439.7) | ||||
| вычислено, %: | 54.63; | 11.23; | 9.55; | 24.6. |
(NBu4)[2-B10H9NC(CH2)3CN] (2). ИК-спектр (CHCl3, см–1): 2487 ν(B–H), 2327 ν(C≡N (B–N)), 2249 ν(C≡N), 1029 δ(B–B–H); 11B ЯМР (CD3CN, δ, м.д.): –0.8 (д, 1B, B(10), JB–H = 146 Гц), –3.5 (д, 1B, B(1), JB–H = 153 Гц), –22.9 (с, 1B, B(2)), –27.5 (д, 3B, B(4,7,8), JB–H = 120 Гц), –29.8 (д, 4B, B(3,5,6,9), JB–H = 120 Гц); 1Н ЯМР (CD3CN, δ, м.д.): –1.01–1.55 (м, 9Н, В10Н9), 3.09 (м, 8H, NBu4), 2.92 (т, 2H, B–NC–CH2–CH2–CH2–CN, J = 8 Гц), 1.98 (квин., 2H, B–NC–CH2–CH2–CH2–CN, J = 8 Гц), 1.60 (м, 8H, NBu4), 1.34 (м, 8H, NBu4), (т, 2H, B–NC–CH2–CH2–CH2–CN, J = 7 Гц), 0.96 (м, 12H, NBu4); 13С ЯМР (CD3CN, δ, м.д.): 119.8 (B–NC–CH2–CH2–CH2–CN), 119.2 (B–NC–CH2–CH2–CH2–CN), 59.3 (NBu4), 24.3 (NBu4), 22.4 (B–NC–CH2–CH2–CH2–CN), 21.3 (B–NC–CH2–CH2–CH2–CN), 20.3 (NBu4), 18.6 (B–NC–CH2–CH2–CH2–CN), 27.4 (B–NC–CH2–CH2–CN), 13.8 (NBu4).
| C | H | N | B | |
|---|---|---|---|---|
| Найдено, %: | 55.80; | 11.82; | 9.25; | 23.1. |
| Для C21H51B10N3 (М = 453.8) | ||||
| вычислено, %: | 55.59; | 11.33; | 9.26; | 23.8. |
(NBu4)[2-B10H9NC(CH2)4CN] (3). ИК-спектр (CHCl3, см–1): 2481 ν(B–H), 2323 ν(C≡N (B–N)), 2258 ν(C≡N), 1031 δ(B–B–H); 11B ЯМР (CD3CN, δ, м.д.): –0.9 (д, 1B, B(10), JB–H = 149 Гц), –3.7 (д, 1B, B(1), JB–H = 151 Гц), –23.1 (с, 1B, B(2)), –27.6 (д, 3B, B(4,7,8), JB–H = 107 Гц), –30.0 (д, 4B, B(3,5,6,9), JB–H = 128 Гц); 1Н ЯМР (CD3CN, δ, м.д.): –1.01–1.55 (м, 9Н, В10Н9), 3.09 (м, 8H, NBu4), 2.86 (т, 2H, B–NC–CH2–CH2–CH2–CH2–CN, J = = 6.42 Гц), 2.43 (м, 4H, B–NC–CH2–CH2–CH2–CH2–CN, J = 6.79 Гц), 1.71 (м, 2H, B–NC–CH2–CH2–CH2–CH2–CN, J = 7.89 Гц), 1.60 (м, 8H, NBu4), 1.34 (м, 8H, NBu4), 1.11 (т, 2H, B–NC–CH2–CH2–CH2–CN, J = 6 Гц), 0.95 (m, 12H, NBu4); 13С ЯМР (CD3CN, δ, м.д.): 120.1 (B–NC–CH2–CH2–CH2–CH2–CN), 116.8 (B–NC–CH2–CH2–CH2–CH2–CN), 59.3 (NBu4), 24.6 (B–NC–CH2–CH2–CH2–CH2–CN), 24.4 (NBu4), 24.1 (B–NC–CH2–CH2–CH2–CH2–CN), 20.3 (NBu4), 18.7 (B–NC–CH2–CH2–CH2–CH2–CN), 17.0 (B–NC–CH2–CH2–CH2–CH2–CN), 13.8 (NBu4).
| C | H | N | B | |
|---|---|---|---|---|
| Найдено, %: | 56.28; | 11.43; | 8.95; | 23.3. |
| Для C22H53B10N3 (М = 439.7) | ||||
| вычислено, %: | 56.49; | 11.42; | 8.98; | 23.1. |
(NBu4)[2-B10H9NCC10H15] (4). ИК-спектр (CHCl3, см–1): 2487 ν(B–H), 2311, ν(C≡N), 1031 δ(B–B–H); 11B ЯМР (CD3CN, δ, м.д.): –0.1 (д, 1B, B(10), JB–H = 148 Гц), –3.0 (д, 1B, B(1), JB–H = 146 Гц), –22.5 (с, 1B, B(2)), –26.4 (д, 3B, B(4,7,8), JB–H = = 115 Гц), –28.9 (д, 4B, B(3,5,6,9), JB–H = 132 Гц); 1Н ЯМР (CD3CN, δ, м.д.): –1.01–1.55 (м, 9Н, В10Н9), 3.15 (м, 8H, NBu4), 2.1–1.6 (м, 15Н, NCC10H15), 1.60 (м, 8H, NBu4), 1.35 (м, 8H, NBu4), 0.95 (м, 12H, NBu4); 13С ЯМР (CD3CN, δ, м.д.): 119.7 (B–NC–Ad), 59.4 (NBu4), 40.4 (Ad, C(2,8,9)), 36.3 (Ad, C(4,6,10)), 32.6 (Ad, C(1)), 27.4 (Ad, C(3,5,7)), 24.5 (NBu4), 20.4 (NBu4), 14.0 (NBu4).
| C | H | N | B | |
|---|---|---|---|---|
| Найдено,%: | 62.38; | 11.82; | 5.35; | 20.2. |
| Для C27H60B10N2 (М = 520.9) | ||||
| вычислено,%: | 62.26; | 11.61; | 5.38; | 20.7. |
(NBu4)[B12H11NCCH3] (5). ИК-спектр (CHCl3, см–1): 2479 ν(B–H), 2339, ν(C≡N), 1032 δ(B–B–H); 11B ЯМР (CD3CN, δ, м.д.): –12.1 (с, 1B, B(1)), ‒15.2 (д, 10B, B(2–11), JB–H = 129 Гц), –17.4 (д, 1B, B(12), JB–H = 127 Гц); 1Н ЯМР (CD3CN, δ, м.д.): ‒1.01–1.55 (м, 11Н, В12Н11), 3.09 (м, 8H, NBu4), 2.51 (с, 3H, C–CH3), 1.60 (м, 8H, NBu4), 1.35 (м, 8H, NBu4), 0.96 (т, 12H, NBu4); 13С ЯМР (CD3CN, δ, м.д.): 125.3 (СN–B), 59.3 (NBu4), 24.3 (NBu4), 20.3 (NBu4), 19.2 (CH3–CN–B), 13.8 (NBu4).
| C | H | N | B | |
|---|---|---|---|---|
| Найдено,%: | 51.11; | 11.97; | 6.55; | 30.0. |
| Для C18H50B12N2 (М = 424.3) | ||||
| вычислено,%: | 50.95; | 11.88; | 6.60; | 30.6. |
(NBu4)[B12H11NCC2H5] (6). ИК-спектр (CHCl3, см–1): 2474 ν(B–H), 2343 ν(C≡N), 1025 δ(B–B–H); 11B ЯМР (CD3CN, δ, м.д.): –13.3 (с, 1B, B(1)), ‒15.9 (д, 11B, B(2–12), JB–H = 125 Гц); 1Н ЯМР (CD3CN, δ, м.д.): –1.01–1.65 (м, 11Н, В12Н11), 3.10 (м, 8H, NBu4), 2.90 (м, 2H, C–CH2–CH3, J = 7 Гц), 1.60 (м, 8H, NBu4), 1.35(м, 8H, NBu4), 1.18 (т, 3H, C–CH2–CH3, J = 8 Гц), 0.96 (т, 12H, NBu4); 13С ЯМР (CD3CN, δ, м.д.): 120.1 (СN–B), 59.3 (NBu4), 28.6 (C–CH2–CH3), 24.3 (NBu4), 20.3 (NBu4), 13.8 (NBu4), 11.5 (C–CH2–CH3).
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
На данный момент методы, позволяющие создавать производные общего вида [B10H9NCR]– с простыми алкильными и арильными заместителями, надежно отработаны. Однако введение более сложных органических фрагментов осложнено нежелательными побочными процессами. Для устранения этого ограничения мы предложили проводить взаимодействие между анионом [B10H11]– и органическими нитрилами в условиях микроволнового синтеза. Такой подход позволяет получать целевые вещества в мягких условиях и с высоким выходом. Следует отметить, что не известны случаи использования ранее микроволнового излучения для синтеза производных нитрилов, есть только несколько примеров применения СВЧ-излучения для модификации нитрильных комплексов платины [38, 39] и нитрилиевых производных кластеров бора [33]. Эффективность СВЧ-синтеза связывают со снижением энергии активации реакций различных типов [19].
С помощью разработанного метода получен ряд нитрилиевых производных клозо-декаборатного аниона на основе твердых и высококипящих органических нитрилов (схема 1 ). Реакция протекает в мягких условиях и отличается легкостью выделения конечного продукта. Использование СВЧ-нагревания позволяет достичь достаточной скорости протекания процесса при незначительном избытке органического нитрила. Предложенная методика распространена также на получение известных ранее производных на основе пропио- и бутиронитрила.
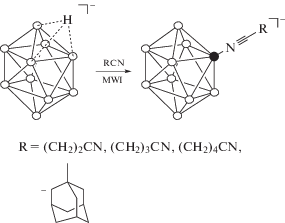
Схема 1 . Общая схема нитрилиевых производных клозо-декаборатного аниона.
Степень протекания процесса оценивали по данным 11B ЯМР-спектроскопии. Так, в 11B ЯМР-спектрах [B10H9NCR]– наблюдаются сигналы от апикальных борных атомов в области –0.1…–0.9 м.д. (I = 1, B(10)) и –2.9…–3.7 м.д. (I = 1, B(1)), сигнал от замещенного атома бора – в области ‒22.5…–23.1 м.д. (I = 1, B(2)), сигналы от незамещенных экваториальных атомов бора – в области –26.6…–27.6 м.д. (I = 3, B(4,7,8)) и –28.9…–30.0 м.д. (I = 4, B(3,5,6,9)). В отсутствие широкополосного подавления спин-спинового взаимодействия сигналы от незамещенных атомов бора проявляются в виде дублетов.
Строение заместителя в полученных соединениях определяли с помощью методов мультиядерной ЯМР-спектроскопии и ИК-спектроскопии поглощения. Так, в 1H ЯМР-спектрах продуктов на основе динитрилов характерными являются сигналы от метиленовых протонов, связанных с активированной тройной связью азот–углерод. Они проявляются в виде триплетов в области 3.22–2.96 м.д. Кроме того, в спектрах продуктов регистрируются сигналы протонов тетрабутиламмониевого катиона и метиленовых групп в β-, γ- и δ-положениях к активированной нитрильной группе. В случае соединения 4 в 1H ЯМР-спектре наблюдаются сигналы протонов адамантильного заместителя в виде серии мультиплетов в области 2.1–1.6 м.д.
В 13C ЯМР-спектрах соединений 1–3 наиболее информативными являются сигналы атомов углерода связанных нитрильных групп в области 126.2–119.8 м.д., сигналы атомов углерода свободных нитрильных групп наблюдаются в области 119.2–116.8 м.д. В 13C ЯМР-спектре соединения 4 сигнал атома углерода нитрильной группы присутствует при 119.7 м.д., в спектре также наблюдаются сигналы атомов адамантильного фрагмента.
Применение микроволнового излучения впервые позволило получить в индивидуальном виде нитрилиевые производные аниона [B12H12]2– (схема 2 ), хотя ранее сообщалось, что ввиду их высокой реакционной способности могут быть выделены только продукты иминольного и амидного типа [40].
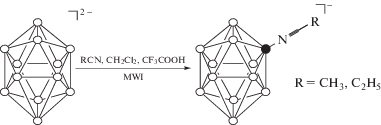
Схема 2 . Общая схема нитрилиевых производных клозо-додекаборатного аниона.
За ходом процесса замещения следили с помощью 11B ЯМР-спектроскопии. Так, в 11B ЯМР-спектре соединения 5 наблюдаются три сигнала: сигнал от замещенного атома бора при –12.1 м.д. (I = 1, B(1)), сигнал от незамещенных экваториальных атомов бора при –15.2 м.д. (I = 10, B(2–11)) и сигнал от атома бора, находящегося в пара-положении к заместителю, при –17.4 м.д. (I = 1, B(12)).
Строение заместителя определяли с помощью методов мультиядерной ЯМР-спектроскопии и ИК-спектроскопии поглощения. В 1H ЯМР-спектре соединения 5 наряду с сигналами протонов тетрабутиламмониевого катиона наблюдается синглет от протона метильной группы при 2.51 м.д. Ацетонитрильная группа в 13С ЯМР-спектре проявляется в виде двух сигналов при 125.3 и 19.2 м.д.
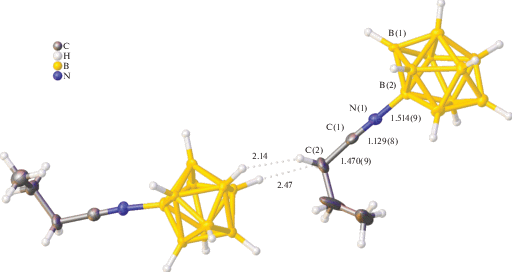
Для соединения (NBu4)[2-B10H9NCnC3H7] структура определена методом рентгеноструктурного анализа (табл. 1, рис. 1). Структура состоит из тетрабутиламмониевых катионов и анионов [2-B10H9NCC3H7]–. Заместитель в анионной части располагается в экваториальном поясе. Экзополиэдрическая связь бор–азот ординарная, ее длина составляет 1.514(9) Å. Параметры связей в нитрилиевом заместителе соответствуют таковым в ранее описанных производных на основе ацетонитрила [15, 18]. Связь N(1)–C(1) незначительно укорочена относительно свободной тройной связи и составляет 1.129(8) Å [41]. Величины углов B(2)N(1)C(1) и N(1)C(1)C(2) составляют 177.8° и 173.6° соответственно. Кроме того, в структуре наблюдается образование коротких межмолекулярных контактов – диводородных связей между атомами α-CH2 заместителя и атомами водорода соседнего кластера (2.14 и 2.47 Å). Наличие этих контактов обусловливает ориентацию анионов в структуре в бесконечные цепи.
Таблица 1.
Кристаллографические данные и параметры уточнения структуры
| Соединение | (NBu4)[2-B10H9NCnC3H7] |
|---|---|
| Эмпирическая формула | C20H52B10N2 |
| М | 428.7 |
| T, K | 100(2) |
| Сингония | Моноклинная |
| Пр. гр. | P21 |
| a, Å | 16.5735(12) |
| b, Å | 20.1342(15) |
| c, Å | 18.1569(12) |
| α, град | 90 |
| β, град | 109.993(5) |
| γ, град | 90 |
| V, Å3 | 3576.87 |
| Z | 8 |
| ρx, мг/м3 | 1.000 |
| μ, мм–1 | 0.052 |
| Размер кристалла, мм | 0.300 × 0.200 × 0.080 |
| Интервал θ, град | 4.10–27.50 |
| Общее число рефлексов независимых (N) [Rint], в том числе с I > 2σ(I) (N0) |
45207 24864 [0.1206] 10994 |
| Tmax, Tmin | – |
| Данные/ограничения/параметры | 24864/1/1153 |
| GOOF (F2) | 0.952 |
| R1, wR2 для N0 | 0.2062, 0.1855 |
| R1, wR2 для N | 0.0871, 0.1374 |
| Δρmax/Δρmin, e/Å3 | 0.238/–0.277 |
ЗАКЛЮЧЕНИЕ
Предложен метод синтеза нитрилиевых производных кластерных анионов бора, основанный на использовании микроволнового излучения. Впервые получены производные клозо-декаборатного аниона на основе динитрилов, а также выделены в индивидуальном виде производные [B12H11NCCH3]– и [B12H11NCC2H5]–.
Список литературы
Xu R. // Introduction – frontiers in modern inorganic synthetic chemistry. Elsevier B.V., 2011. https://doi.org/10.1016/B978-0-444-53599-3.10001-0
Shmal’ko A.V., Sivaev I.B. // Russ. J. Inorg. Chem. 2019. V. 64. № 14. P. 1726. https://doi.org/10.1134/S0036023619140067
Stockmann P., Gozzi M., Kuhnert R. et al. // Chem. Soc. Rev. 2019. V. 48. № 13. P. 3497. https://doi.org/10.1039/c9cs00197b
Fisher S.P., Tomich A.W., Lovera S.O. et al. // Chem. Rev. 2019. V. 119. № 14. P. 8262. https://doi.org/10.1021/acs.chemrev.8b00551
Quan Y., Xie Z. // Chem. Soc. Rev. 2019. V. 48. № 13. P. 3660. https://doi.org/10.1039/c9cs00169g
Kiremire E.M.R. // Int. J. Chem. 2016. V. 8. № 3. P. 62. https://doi.org/10.5539/ijc.v8n3p62
Hansen B.R.S., Paskevicius M., Li H.W. et al. // Coord. Chem. Rev. 2016. V. 323. P. 60. https://doi.org/10.1016/j.ccr.2015.12.003
Sivaev I.B., Prikaznov A.V., Naoufal D. // Collect. Czechoslov. Chem. Commun. 2010. V. 75. № 11. P. 1149. https://doi.org/10.1135/cccc2010054
Zhizhin K.Y., Zhdanov A.P., Kuznetsov N.T. // Russ. J. Inorg. Chem. 2010. V. 55. № 14. P. 2089. https://doi.org/10.1134/S0036023610140019
Zhdanov A.P., Voinova V.V., Klyukin I.N. et al. // J. Clust. Sci. 2019. V. 2. https://doi.org/10.1007/s10876-019-01628-2
Kubasov A.S., Matveev E.Y., Turyshev E.S. et al. // Inorg. Chim. Acta. 2018. V. 477. P. 277. https://doi.org/10.1016/j.ica.2018.03.013
Burianova V.K., Bolotin D.S., Mikherdov A.S. et al. // New J. Chem. 2018. V. 42. № 11. P. 8693. https://doi.org/10.1039/c8nj01018h
Burianova V.K., Bolotin D.S., Novikov A.S. et al. // Inorg. Chim. Acta. 2018. V. 482. P. 838. https://doi.org/10.1016/j.ica.2018.07.038
Bolotin D.S., Burianova V.K., Novikov A.S. et al. // Organometallics. 2016. V. 35. № 20. P. 3612. https://doi.org/10.1021/acs.organomet.6b00678
Dou D., Mavunkal I.J., Bauer J.A.K. et al. // Inorg. Chem. 1994. V. 33. № 26. P. 6432. https://doi.org/10.1021/ic00104a069
Zhdanov A.P., Nelyubin A.V., Klyukin I.N. et al. // 2019. V. 64. № 7. P. 841. https://doi.org/10.1134/S0036023619070180
Nelyubin A.V., Klyukin I.N., Zhdanov A.P. et al. // Russ. J. Inorg. Chem. 2019. V. 64. № 14. P. 1750. https://doi.org/10.1134/S0036023619140043
Жижин К.Ю., Мустяца В.Н., Матвеев Е.Ю. и др. // Журн. неорган. химии. 2003. V. 48. № 5. P. 760.
André L. // Microwaves in Organic Synthesis, Second Edi, Weinheim, 2006. https://doi.org/10.1002/9783527619559
Zhu Y.J., Chen F. // Chem. Rev. 2014. V. 114. № 12. P. 6462. https://doi.org/10.1021/cr400366s
Takkellapati S. // Curr. Org. Chem. 2013. V. 17. № 20. P. 2305. https://doi.org/10.2174/13852728113179990042
Abe T., Miyazawa A., Kawanishi Y. et al. // Mini-Rev. Org. Chem. 2011. V. 8. № 3. P. 315. https://doi.org/10.2174/157019311796197346
Schwenke A.M., Hoeppener S., Schubert U.S. // Adv. Mater. 2015. V. 27. № 28. P. 4113. https://doi.org/10.1002/adma.201500472
Yapryntsev A.D., Bykov A.Y., Baranchikov A.E. et al. // Inorg. Chem. 2017. V. 56. № 6. P. 3421. https://doi.org/10.1021/acs.inorgchem.6b02948
Nakamura H., Tasaki L., Kanoh D. et al. // Dalton Trans. 2014. V. 43. № 13. P. 4941. https://doi.org/10.1039/c3dt52828f
Gona K.B., Thota J.L.V.N.P., Baz Z. et al. // Dalton Trans. 2015. V. 44. № 21. P. 9915. https://doi.org/10.1039/c5dt01049g
Armstrong A.F., Valliant J.F. // Inorg. Chem. 2007. V. 46. № 6. P. 2148. https://doi.org/10.1021/ic0617543
Wixtrom A.I., Shao Y., Jung D. et al. // Inorg. Chem. Front. 2016. V. 3. № 5. P. 711. https://doi.org/10.1039/c5qi00263j
Kamin A.A., Juhasz M.A. // Inorg. Chem. 2020. V. 59. № 1. P. 189. https://doi.org/10.1021/acs.inorgchem.9b03037
Valášek M., Štursa J., Pohl R. et al. // Inorg. Chem. 2010. V. 49. № 22. P. 10247. https://doi.org/10.1021/ic101234p
Hsu M.H., Hsieh C.Y., Kapoor M. et al. // Bioorg. Chem. 2020. V. 98. № December. 2019. P. 103729. https://doi.org/10.1016/j.bioorg.2020.103729
Dopke J.A., Lincoln Z.S., Blazejewski J. et al. // Inorg. Chim. Acta. 2018. V. 473. P. 263. https://doi.org/10.1016/j.ica.2017.12.027
Ezhov A.V., Vyal’ba F.Y., Kluykin I.N. et al. // Macroheterocycles. 2017. V. 10. № 4–5. P. 505. https://doi.org/10.6060/mhc171254z
SAINT. Version 7.23A (Bruker, Madison, WI, 2003)
SADABS-2004/1 (Bruker, Madison, WI, 2003).
Sheldrick G.M. // Acta Crystallogr. A. 2008. V. 64. P. 112. https://doi.org/10.1107/S0108767307043930
Sheldrick G.M. // Acta Crystallogr., Sect. A: Found. Crystallogr. 2015. V. 71. № 1. P. 3. https://doi.org/10.1107/S2053273314026370
Charmier M.A.J., Kukushkin V.Y., Pombeiro A.J.L. // Dalton Trans. 2003. № 12. P. 2540. https://doi.org/10.1039/b301892j
Luzyanin K.V., Kukushkin V.Y., Kuznetsov M.L. et al. // Inorg. Chem. 2002. V. 41. № 11. P. 2981. https://doi.org/10.1021/ic025554c
Stogniy M.Y., Erokhina S.A., Sivaev I.B. et al. // Phosphorus, Sulfur Silicon Relat. Elem. 2019. V. 194. № 10. P. 983. https://doi.org/10.1080/10426507.2019.1631312
Allen F.H., Kennard O., Watson D.G. et al. // J. Chem. Soc., Perkin Trans. II. 1987. P. S1. https://doi.org/10.1039/p298700000s1
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии