

Журнал неорганической химии, 2021, T. 66, № 12, стр. 1739-1747
Теоретическое моделирование реакций экзо- и эндоэндрального гидрирования допированного магниевого кластера Mg17Ni
А. П. Мальцев a, b, *, О. П. Чаркин a, **
a Институт проблем химической физики РАН
142432 Черноголовка, Московская обл., пр-т Академика Семенова, 1, Россия
b Московский государственный университет имени М.В. Ломоносова
119991 Москва, Ленинские горы, 1, Россия
* E-mail: maltsevaleksej@gmail.com
** E-mail: charkin@icp.ac.ru
Поступила в редакцию 20.04.2021
После доработки 01.06.2021
Принята к публикации 27.06.2021
Аннотация
На основании DFT- и IRC-расчетов сопоставлены поверхности потенциальной энергии каналов гидрирования кластера Mg17Ni. В “экзоэдральных” каналах сорбционные, постсорбционные и миграционные процессы атомов H полагаются локализованными на поверхности каркаса с максимальными барьерами ~14 ккал/моль. В “эндоэдральном” канале с барьером ~6 ккал/моль ключевую роль играют трансформации плоской конфигурации поверхностной NiH4-группы в тетраэдрическую, при которой два ее нижних атома Н мигрируют через внутреннюю полость и выходят на поверхностные позиции, инвертируя через квадратную и смежные треугольные грани в нижней половине каркаса. Протекание последнего процесса требует более мягких условий по сравнению с экзоэдральными каналами.
ВВЕДЕНИЕ
Магний является перспективным материалом для обратимого хранения водорода благодаря высокой емкости образующихся гидридов, однако его применение ограничено из-за ряда недостатков, к которым относятся высокие активационные барьеры сорбции-десорбции водорода и медленные скорости диффузии водорода через гидридный слой [1–3]. Эффективным способом улучшения кинетических характеристик считается введение каталитических добавок, таких как переходные 3d-металлы, и изменение микроструктуры сплавов с увеличением площади межфазных границ и уменьшением размеров зерен до субмикронных или наноразмерных [4, 5]. Тем не менее достигнутые результаты еще не полностью соответствуют требованиям для практического применения магниевых сплавов, а экспериментальное изучение наноразмерных материалов на основе магния, в том числе механизмов их гидрирования, сопровождается техническими трудностями. В связи с этим в последние годы быстро растет интерес к квантово-химическому изучению наноразмерных магниевых кластеров и моделированию элементарных процессов сорбции-десорбции водорода на металлических кластерах или магниевых поверхностях.
Благодаря интенсивному развитию методов масс-спектроскопии, ИК-спектроскопии в низкотемпературных инертных матрицах в совокупности с применением квантово-химических расчетов, стало возможным получать и идентифицировать наноразмерные смешанные магний-алюминиевые кластеры [6–10], гомоядерные магниевые кластеры [11] и их гидриды [12]. В последние годы активно проводятся квантово-химические расчеты гомоядерных магниевых кластеров Mgn (n = 2–56) [13–18], особенно стремительно растет интерес к квантово-химическим расчетам малых магниевых кластеров. Так, в работах [19–21] проведены расчеты серии бериллий-допированных магниевых кластеров ${\text{M}}{{{\text{g}}}_{n}}{\text{Be}}_{m}^{Q}$ (n = 1–20; m = 1, 2; Q = 0, –1), найдены глобальные минимумы структур, определены их энергетические характеристики, установлено, что наиболее стабильными являются кластеры BeMg9, Be2Mg5, ${\text{B}}{{{\text{e}}}_{2}}{\text{Mg}}_{5}^{ - }$ и ${\text{B}}{{{\text{e}}}_{2}}{\text{Mg}}_{8}^{ - }$. Аналогичные расчеты выполнены для кластеров MgnSi (n = 3–12) [22], MgnZnm (n = 1–5, m = 1, 2) [23], MgnZr (n = 1–11) [24], Mg2Bn (n = 4–14) [25], MgnX (X = Ge, C, Sn; n = 2–12) [26]. Для кластеров MgnZnm и Mg2Bn авторами работ [23 и 25] рассчитаны соответственно гидриды MgnZnmH2 и Mg2BnH2, определены их геометрические и энергетические параметры. Показано, что энергия хемосорбции в кластерах, допированных Zr, варьируется от 20 до 30 ккал/моль, а энергия физической сорбции в кластерах Mg2Bn составляет ~5 ккал/моль.
Значительный интерес представляет квантово-химическое моделирование элементарных реакций гидрирования допированных кластеров. Ранее эта проблема подробно исследовалась для родственных реакций наноразмерных алюминиевых кластеров, для которых были рассчитаны поверхности потенциальной энергии (ППЭ) вдоль минимальных энергетических путей (МЭП) с участием допантов Mg, Si [27, 28], B, C, P, Ca [28] и атомов переходных металлов 3d- и 4d-периодов [29–32]. Реакции водорода с допированными магниевыми кластерами исследованы в меньшей степени. В работе [33] выполнены расчеты ППЭ гидрирования магний-алюминиевого кластера Mg5Al7Ni, а в работах [34, 35] – допированных магниевых кластеров MgnL (n = 2–10; M = Rh, Co), показано, что энергия присоединения молекулы H2 составляет ~15–20 ккал/моль.
Недавно проведены расчеты ППЭ реакций гидрирования гомоядерного кластера Mg18 [36], допированных кластеров Mg17L
(1)
${\text{M}}{{{\text{g}}}_{{{\text{17}}}}}{\text{L}} + {{{\text{H}}}_{2}} \to {\text{M}}{{{\text{g}}}_{{{\text{17}}}}}{\text{L}}{{{\text{H}}}_{{\text{2}}}}$Рис. 1.
Оптимизированные ключевые структуры интермедиатов и переходных состояний каталитического цикла реакций (1).
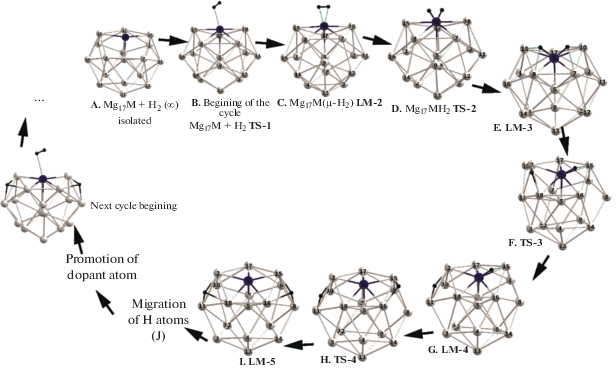
Согласно [36], барьеры перемещений атомов Н на поверхности кластера Mg18 не превышают нескольких ккал/моль и свидетельствуют о возможности квазисвободной поверхностной миграции атомов Н у родственных гомоядерных кластеров в условиях обычных или умеренно повышенных температур. Энергия гидрирования кластера Mg18 оценивается в ~5 ккал/моль, но осуществление этой реакции затруднено высоким активационным барьером сорбции и диссоциации молекулы Н2, достигающим ~30 ккал/моль. Замещение поверхностного атома Mg атомом L переходного 3d-металла снижает этот барьер до ~1–3 ккал/моль, так что в системах с допантами 3d-элементов сорбция Н2 и образование “первичного” дигидрида Mg17LH2 (E) должны проходить без существенных изменений энергии. Энергии образования Mg17LH2 (E) монотонно уменьшаются с увеличением порядкового номера допанта L от ~30 и ~20 ккал/моль у Ti и V до ~12 ккал/моль у Ni. В постсорбционном участке реакции лимитирующей является стадия очистки допанта М от связанных с ним атомов H. Соответствующие ей активационные барьеры тоже уменьшаются с увеличением порядкового номера допанта L от ~20 ккал/моль у Ti и V до ~10–12 ккал/моль у Ni [37, 38].
При последовательном присоединении первых шести (n = 6) молекул Н2 к кластеру Mg17Ni (параллельно со смещениями атомов Н по поверхности) атом допанта тоже смещается с поверхностной позиции и погружается (“ныряет”) во внутреннюю полость каркаса Mg17 на ~1.5 Å и более. В подобных “закрытых” внутренних позициях допант экранирован соседними атомами магния, затрудняющими его взаимодействие с новой молекулой Н2. Для восстановления каталитической активности допант должен быть возвращен (“промотирован”) в открытое поверхностное положение. Оценки необходимых для этого энергий промотирования (Епром) не превышают умеренных 4–6 ккал/моль у первых трех реакций (n = 3), но увеличиваются до ~20 ккал/моль при n = 6, так что промотирование становится лимитирующей стадией при увеличении n и может привести к обрыву цепочки циклов при скромном числе сорбированных молекул Н2. При значительных n картина еще усложняется одновременными структурными перестройками каркаса Mg17, в ходе которых один или несколько атомов Mg выходят из состава каркаса в “надповерхностную” сферу и образуют внешние терминальные связи Ni−Mg, которые еще сильнее экранируют допант и тоже могут обрывать процесс последовательного гидрирования.
Настоящая работа является продолжением работ [36–38] и посвящена поиску способов обойти трудности, связанные c энергиями промотирования допанта, с использованием модифицированного цикла
(2)
${{{\text{H}}}_{{2n}}}_{{--2}}{\text{M}}{{{\text{g}}}_{{17}}}\left( {{\text{Ni}}{{{\text{H}}}_{2}}} \right) + {{{\text{H}}}_{2}} \to {{{\text{H}}}_{{2n}}}{\text{M}}{{{\text{g}}}_{{17}}}\left( {{\text{Ni}}{{{\text{H}}}_{2}}} \right),$Цель настоящей работы – расчет геометрических, энергетических и электронных характеристик ключевых структур, отвечающих особым точкам ППЭ серии реакций (2), и сравнение относительных энергий интермедиатов и активационных барьеров реакций (2) с аналогичными характеристиками реакций (1) гидрирования голого кластера Mg17Ni [36].
РАСЧЕТНЫЕ МЕТОДЫ
Как и ранее [36–38], расчеты выполняли в рамках метода функционала плотности (DFT) BP86 [39] c использованием программы GAUSSIAN-09 [40] и базиса 6-31G*. Характер особых точек ППЭ контролировали расчетами частот нормальных колебаний и методом внутренней координаты реакции (irc). Согласно нашим DFT-расчетам синглетного и триплетного термов, выполненным ранее в [37], у всех локальных минимумов и переходных состояний вдоль рассмотренных минимальных энергетических путей равновесные структуры у обоих термов очень близки, а триплет лежит выше синглета на почти постоянную величину ~10 ккал/моль.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Реакция H2nMg17(NiH2) + H2 → H2n+2Mg17(NiH2)
Начальный (сорбционный) участок ППЭ реакции (1), отвечающий присоединению первой молекулы Н2 к голому кластеру Mg17Ni и образованию первичного дигидрида Mg17NiH2 (Е), ранее подробно рассматривался в [36]. На рис. 2 и 3 приведены ключевые структуры, отвечающие МЭП реакции
(2а)
${\text{M}}{{{\text{g}}}_{{17}}}({\text{Ni}}{{{\text{H}}}_{2}}) + {{{\text{H}}}_{2}} \to {{{\text{H}}}_{{\text{2}}}}{\text{M}}{{{\text{g}}}_{{{\text{17}}}}}{\text{(Ni}}{{{\text{H}}}_{2}}),$Рис. 2.
Оптимизированные ключевые структуры интермедиатов и переходных состояний реакции Mg17NiH2 + H2 → → H2Mg17(NiH2) (2а).
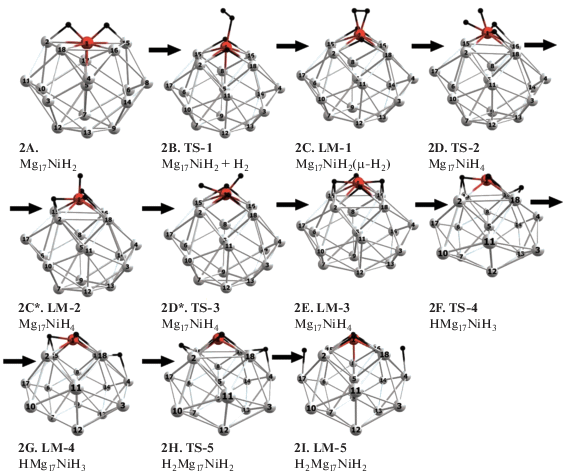
Рис. 3.
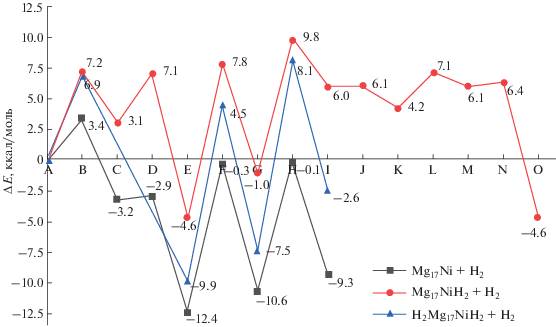
Энергии ключевых структур интермедиатов и переходных состояний на первых трех циклах гидрирования Mg17Ni + H2 и H2n–2Mg17NiH2 + H2. Структуры и диаграммы реакции Mg17Ni + H2 → Mg17NiH2 (1) приведены в [36].
и молекулярные диаграммы
(2б)
${{{\text{Н}}}_{{\text{2}}}}{\text{M}}{{{\text{g}}}_{{{\text{17}}}}}{\text{(Ni}}{{{\text{H}}}_{2}}) + {{{\text{H}}}_{2}} \to {{{\text{H}}}_{{\text{4}}}}{\text{M}}{{{\text{g}}}_{{{\text{17}}}}}{\text{(Ni}}{{{\text{H}}}_{2}}).$На стадии сорбции присоединения второй молекулы Н2 к дигидриду Mg17NiH2 (E) образуется тетрагидрид Mg17NiH4 (2E) с группой NiH4, расположенной над поверхностью магниевого каркаса на расстоянии ~1.23 Å. Дигидрид Е и тетрагидрид 2E разделены двумя барьерами ~7 ккал/моль, первый (2В) из которых отвечает физической сорбции молекулы Н2 и образованию комплекса μ-Н2, а последний – диссоциации молекулы водорода на атомы. Из сравнения молекулярных диаграмм на рис. 3 можно проследить ряд отличий реакций (1) и (2) на стадии сорбции. У реакции (2а) барьер физосорбции (2В) вдвое выше, чем у реакции (1). У реакции (1) диссоциация молекулы Н2 с образованием дигидрида Mg17NiH2 проходит в одну стадию (при одновременном образовании обоих симметричных мостиков Ni−Hb−Mg) c малоощутимым барьером (несколько десятых долей ккал/моль), в то время как у реакции (2а) распад Н2 на атомы протекает в две ступени с последовательным образованием первого и второго водородных мостиков, причем барьер диссоциации 2D вполне ощутим и сопоставим с барьером физосорбции 2Е (~7 ккал/моль). Кроме того, в отличие от реакции (1), при переходе от дигидрида Е к тетрагидриду 2Е реакция (2а) проходит через интермедиат NiH3H (2С*), в котором три атома H расположены на смежных гранях Mg–Mg–Ni, а четвертый атом Н образует вертикальную терминальную связь NiH (рис. 2). Комплекс NiH3H (2С*) отделен от тетрагидрида 2Е небольшим барьером 2D* ~1.5 ккал/моль.
Внутри сорбционного участка 2B–2C реакции (2а) имеет место перестройка структуры магниевого каркаса, которая приближается к структуре сферически ароматичного кластера Mg17 [16] и с небольшими деформациями сохраняется на остальных стадиях (от Е до J) не только у реакции (2а), но и в последующих реакциях присоединения третьей и четвертой молекул Н2. По сравнению с дигидридом Mg17NiH2 (E) длины связей Mg–Mg в тетрагидриде Mg17NiH4 (2Е) укорачиваются на 0.05–0.1 Å, а их колебательные частоты увеличиваются на 5–10 см–1. В квадратной группе NiH4 тетрагидрида Mg17NiH4 (2Е) атомы Н располагаются в мостиковых позициях над магниевыми ребрами с расстояниями R(NiH) = 1.56 и R(MgH) = 2.08 Å и углом φ(MgHMg) = 100°. Энергия образования Mg17NiH4 (2Е) при присоединении Н2 к дигидриду Mg17NiH2 (Е) оценивается в ~4.6 ккал/моль.
Как и у реакции (1), по завершении сорбционной стадии и образовании тетрагидрида в реакции (2а) реализуется стадия “очистки допанта” за счет перемещения присоединившихся атомов Н к удаленным граням или ребрам магниевого каркаса и образования интермедиатов 2G и 2I'. Активационные барьеры 2F и 2H, разделяющие интермедиаты 2E, 2G и 2I, составляют 12.4 и 10.8 ккал/моль соответственно и сопоставимы с аналогичными барьерами 1F и 1H, рассчитанными в [36], в пределах нескольких ккал/моль. При сравнении диаграмм реакций (1) и (2), приведенных на рис. 3, можно видеть их подобие и полуколичественное сходство как в структурах, так и в положении ключевых структур на энергетической шкале. У обеих реакций лимитирующей остается стадия очистки допанта. Количественные отличия проявляются в том, что сорбционный барьер 2В на 3.5 ккал/моль выше барьера 1В, а постсорбционные барьеры 2F и 2H тоже на несколько ккал/моль выше аналогов 1F и 1H. Структура 2I лежит на 6 ккал/моль выше изолированной системы Mg17NiH2 + H2 и на 6 ккал/моль ниже диссоциационного предела Mg17Ni + 2H2. В отличие от (1), в реакции (2) допант везде остается фиксированным на “открытой” поверхностной позиции в составе группы NiH2 и в структуре 2I готов к присоединению следующей молекулы Н2 без затрат энергии на промотирование. Та же качественная картина с допантом, фиксированным на поверхностной позиции в структуре 3I, получена в наших расчетах аналогичного цикла (2б), в котором к дигидридной группе NiH2 присоединяется третья молекула Н2 и т. д.
Рис. 4.
Оптимизированные ключевые структуры интермедиатов и переходных состояний каталитического цикла гидрирования H2Mg17NiH2 + H2. Стадии поверхностной миграции изображены на рисунке S1 приложения.
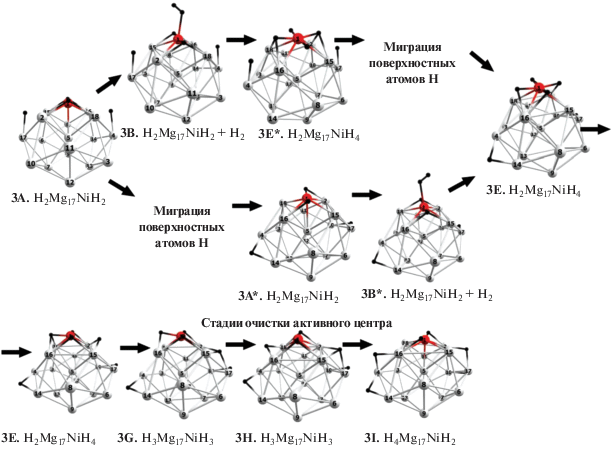
В реакции (2б) двухэтапное гидрирование структуры H2Mg17NiH2 (2I) в тетрагидрид H2Mg17NiH4 может осуществляться двумя путями (рис. 4). Первый путь (верхний на рис. 4) начинается с сорбции третьей молекулы Н2 и образования интермедиата 3E* с группой NiH4, в котором удаленные атомы Н локализованы в экваториальном поясе каркаса. На постсорбционном этапе удаленные атомы Н смещаются из экватора в нижнюю полусферу каркаса с образованием более выгодной структуры тетрагидрида 3Е. На втором пути (нижнем на рис. 4) меняется последовательность этапов: он начинается с перемещения атомов Н из экватора в нижнюю полусферу с образованием интермедиата H2Mg17(NiH2) (3E**), в котором сохраняется группа NiH2, и заканчивается сорбцией третьей молекулы Н2 c образованием того же тетрагидрата 3Е. Обоим каналам отвечают низкие потенциальные барьеры.
Поверхностная миграция атомов Н проходит через пологие локальные минимумы 2K и 2М, отделенные от I и друг от друга барьерами 2J, 2L и 2N (рис. S1 ), высота которых не превышает 3 ккал/моль. На последнем участке 2M–2N–2О смещение атома Н оказывается связанным с переориентацией группы NiH2 и структурной перестройкой каркаса, которые сопровождаются суммарным выигрышем энергии интермедиата 2О по сравнению с 2I на ~10 ккал/моль (рис. 3). К такому же выигрышу энергии приводит сорбция третьей молекулы водорода и образование тетрагидрида H2Mg17NiH4 (Е) относительно изолированных реагентов H2Mg17NiH2 + H2. На первом пути он отвечает стадии сорбции, а на втором – сложной кооперативной перегруппировке, включающей одновременно миграцию атомов Н, поворот группы NiH2 и перестройку каркаса Mg17.
Продолжение цикла с присоединением новых молекул водорода может следовать той же схеме аналогично рассмотренным выше циклам с активным центром NiH2. Их ключевой стадией должна остаться очистка допанта с перемещениями пары атомов Н из группы NiH4 в предпочтительные поверхностные позиции 3G и 3Ι в экваторе каркаса, которые разделены друг от друга и от тетрагидрида 3E активационными барьерами 3F и 3H ~14–16 ккал/моль.
Реакции “эндоэдрального” гидрирования
Обсуждаемые выше каналы относятся к реакциям “поверхностного гидрирования”, в которых группа NiH4 имеет квазиплоское строение и в которых перемещения и миграция присоединившихся атомов Н происходят на поверхности кластера. У всех таких реакций очистка допанта от атомов Н оказывается лимитирующей стадией c активационными барьерами F и Н, которые нигде не опускаются ниже ~10–14 ккал/моль и служат препятствием для проведения этих реакций в мягких условиях.
Рассмотрим теперь альтернативный канал “эндоэдрального” гидрирования, в котором атомы водорода перемещаются с поверхности вглубь каркаса и мигрируют в его внутренней полости. Этот канал можно условно разбить на несколько стадий. Сначала один из присоединившихся атомов (ниже Н*) уходит с поверхности внутрь каркаса и включается в состав эндоэдрального мостика Ni−H*−Mgc между допантом и центральным атомом Mgc. После этого атом Н* удаляется от допанта и мигрирует в нижнюю половину каркаса через систему промежуточных мостиков между атомом Mgc и ребрами или гранями каркаса. Далее атом Н* инвертирует через нижнее квадратное окно в каркасе, находящееся в транс-положении к допанту, выходит из внутренней полости на поверхность, занимает позицию над прилежащим ребром или гранью и далее может мигрировать обычным образом на другие поверхностные позиции кластера. Ключевые структуры вдоль одного из путей миграции и их относительные энергии (в ккал/моль) приведены на рис. 5. Более полный список структур и их полные энергии представлены на рис. S2 .
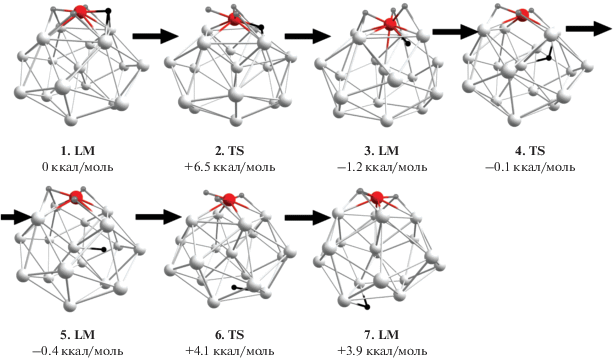
Согласно расчетам, структура 1 с плоской NiH4-группой не является энергетически предпочтительной. На ~1.2 ккал/моль ниже ее лежит интермедиат 3 с тетраэдрической группой НNiH3, который отделен от 1 “входным” барьером ~6.5 ккал/моль. В ходе перегруппировки NiH4 → HNiH3 между этими плоской и тетраэдрической формами три внешних атома Н остаются координированными к верхним связям Ni−Mg, а четвертый атом Н* инвертирует через смежное ребро Mg−Mg, растягивающееся при этом почти на 1 Å, опускается внутрь каркаса и включается в эндоэдральный треугольник NiHMgc c расстояниями R(NiH) = 1.57, R(MgcH) = = 2.10 и R(NiMgc) = 2.78 Å. Интермедиат 3 интересен тем, что, за исключением области, близкой к допанту, его ППЭ внутренней миграции атома Н в лежащие ниже позиции представляет собой совокупность мелких локальных минимумов, разделенных низкими барьерами (не выше 1.5–2 ккал/моль), которые способствуют квазисвободной миграции Н* при обычных или слегка повышенных температурах.
Аналогичные результаты получаются в расчетах последовательной инверсии обоих противолежащих атомов H плоской группы NiH4 с образованием тетраэдрического изомера H2NiH2, в котором пара верхних атомов остается фиксированной в составе группы NiH2, а два атома Н перемещаются во внутреннюю полость с образованием эндоэдрального ромба Ni(H)2Mgc. В перегруппировке NiH4 → H2NiH2 (ее механизм cм. на рис. S3 и S4 приложения) последние два атома Н последовательно инвертируют через растягивающиеся смежные ребра примерно с одинаковым барьером ~6.5 ккал/моль на каждую инверсию, близким к барьеру перегруппировки NiH4 → HNiH3. В обоих случаях этот входной инверсионный барьер оказывается вдвое ниже барьеров G и H очистки допанта в реакциях (1) и (2). Отметим, что, в отличие от вполне ощутимого входного барьера при трансформации плоской NiH4-группы в тетраэдрическую, выходной инверсионный барьер ~4 ккал/моль представляет меньше трудностей для миграции атомов Н из внутренней полости на поверхность через нижнее “окно” каркаса. По мере выхода атомов Н из полости к группе NiH2 может присоединиться новая молекул Н2 с образованием плоской группы NiH4 и началом нового цикла. Можно полагать, что эндоэдральное гидрирование будет проходить в более мягких условиях, чем экзоэдральное. Можно также полагать, что эндоэдральный канал представляет интерес и для обратного процесса десорбции с легким переходом поверхностных атомов Н во внутреннюю полость через нижнее окно и с умеренным барьером выхода на поверхность через образование плоской группы NiH4 с последующим освобождением молекулы Н2 из комплекса Mg17NiH2(μ-H2).
Отметим, что попытки оценить возможности удлинения цепочек из следующих друг за другом циклов типа (1) или (2) пока преждевременны. По мере увеличения n и заполнения поверхности атомами Н картина может усложняться из-за нарастания искажений каркаса Mg17 с разрывом напряженных ребер Mg−Mg, образованием более крупных 5- и 6-атомных “окон” и появлением новых каналов перемещения атомов Н с поверхности во внутреннюю полость каркаса и обратно. Важность каркасных деформаций и перегруппировок с увеличением n была продемонстрирована ранее на примере последовательно гидрированных алюминиевых кластеров Al44Hn и Al86Hm [41]. Cвязи Mg−Mg менее прочны и более лабильны по сравнению со связями Al−Al, и следует ожидать, что при гидрировании магниевых кластеров “каркасные” эффекты должны проявляться еще более отчетливо. Эти вопросы являются предметом наших дальнейших исследований.
Список литературы
Yartys V.A., Lototskyy M.V., Akiba E. et al. // Int. J. Hydrogen Energy. 2019. V. 44. P. 7809. https://doi.org/10.1016/j.ijhydene.2018.12.212
Tarasov B.P., Arbuzov A.A., Mozhzhuhin S.A. et al. // Int. J. Hydrogen Energy. 2019. V. 45. P. 29212. https://doi.org/10.1016/j.ijhydene.2019.02.033
Ouyang L., Liu F., Wang H. et al. // J. Alloys Compd. 2020. V. 832. P. 154865. https://doi.org/10.1016/j.jallcom.2020.154865
Yao X., Lu G. // Chin. Sci. Bull. 2008. V. 53. P. 2421. https://doi.org/10.1007/s11434-008-0325-2
Huang L.J., Wang H., Liu J.W. et al. // J. Alloys Compd. 2019. V. 792. P. 835. https://doi.org/10.1016/j.jallcom.2019.04.029
Wang H., Ko Y.J., Zhang X. et al. // J. Chem. Phys. 2014. V. 140. P. 124309. https://doi.org/10.1063/1.4869104
Luo Z., Grover C.J., Reber A.C. et al. // J. Am. Chem. Soc. 2013. V. 135. P. 4307. https://doi.org/10.1021/ja310467n
Grover C.J., Reber A.C., Khanna S.N. // J. Chem. Phys. 2017. V. 146. P. 2243301. https://doi.org/10.1063/1.4985093
Lu B.J., Li X.-T., Zhao Y.-J. et al. // AIP Adv. 2017. V. 7. P. 095023. https://doi.org/10.1063/1.5000792
Zhang L., Ma X., Guo X. et al. // J. Clust. Sci. 2020.https://doi.org/10.1007/s10876-020-01803-w
Kaufmann A., Kornath A., Zoermer A. et al. // Inorg. Chem. 2010. V. 49. P. 3851. https://doi.org/10.1021/ic902485z
Wang X., Andrews L. // J. Phys. Chem. A. 2004. V. 108. P. 11511. https://doi.org/10.1021/jp046410h
Janecek S., Krotscheck E., Liebrecht M. et al. // Eur. Phys. J. D. 2011. V. 63. P. 377. https://doi.org/10.1140/epjd/e2011-10694-2
Köhn A., Weigend F., Ahlrichs R. // Phys. Chem. Chem. Phys. 2001. V. 3. P. 711. https://doi.org/10.1039/B007869G
Heidari I., De S., Ghazi S.M. et al. // J. Phys. Chem. A. 2011. V. 115. P. 12307. https://doi.org/10.1021/jp204442e
Xia X., Kuang X., Lu C. et al. // J. Phys. Chem. A. 2016. V. 120. P. 677. https://doi.org/10.1021/acs.jpca.6b07322
Jellinek J., Acioli P.H. // J. Phys. Chem. A. 2002. V. 106. P. 10919. https://doi.org/10.1021/jp020887g
Duanmu K., Roberto-Neto O., Machado F.B.C. et al. // J. Phys. Chem. C. 2016. V. 120. P. 13275. https://doi.org/10.1021/acs.jpcc.6b03080
Zhang F., Zhang H., Xin W. et al. // Scientific Reports. 2020. V. 10. P. 6052. https://doi.org/10.1038/s41598-020-63237-8
Zeng L., Wei X.-F., Liang M.-K. et al. // Comput. Mater. Sci. 2020. V. 182. P. 109795. https://doi.org/10.1016/j.commatsci.2020.109795
Zeng L., Deng P.-J., Bi J., Zhu B.-C. // J. Comput. Chem. 2020.https://doi.org/10.1002/jcc.26359
Zhu B.-C., Zhang S., Zeng L. // Int. J. Quantum. Chem. 2020. V. 120. P. 26143. https://doi.org/10.1002/qua.26143
Li Z., Zhao Z., Zhou Z. et al. // Mater. Chem. Phys. 2020. V. 199 P. 585. https://doi.org/10.1016/j.matchemphys.2017.07.049
He C., Chen Y., Sheng Y. // Eur. Phys. J. D. 2019. V. 73. P. 90. https://doi.org/10.1140/epjd/e2019-90521-6
Kumar A., Vyas N., Ojha A.K. // Int. J. Hydrogen Energy. 2020. V. 45. P. 12961. https://doi.org/10.1016/j.ijhydene.2020.03.018
Zeng L., Liang M.-K., Wei X.-F. et al. // J. Phys.: Condens. Matter. 2020. V. 33. https://doi.org/10.1088/1361-648X/abc401
Henry D.J., Yarovsky I. // J. Phys. Chem. A. 2009. V. 113. P. 2565. https://doi.org/10.1016/10.1021/jp809619q
Wang L., Zhao J., Zhou Z. et al. // J. Comput. Chem. 2009. V. 30. P. 2509. https://doi.org/10.1016/10.1002/jcc.21239
Kochnev V.K., Charkin O.P., Klimenko N.M. // Russ. J. Inorg. Chem. 2010. V. 55. P. 65. https://doi.org/10.1134/S0036023610010134
Guo L. // J. Phys. Chem. 2013. V. 117. P. 3458. https://doi.org/10.1021/jp310833y
Mikhailin A.A., Charkin O.P., Klimenko N.M. // Russ. J. Inorg. Chem. 2013. V. 58. P. 1479. https://doi.org/10.1134/S0036023613120073
Mikhailin A.A., Charkin O.P., Klimenko N.M. // Russ. J. Inorg. Chem. 2016. V. 61. P. 1558. https://doi.org/10.1134/S0036023616120135
Mikhailin A.A., Charkin O.P., Klimenko N.M. // Russ. J. Inorg. Chem. 2015. V. 60. P. 1238. https://doi.org/10.1134/S0036023615100137
Trivedi R., Bandyopadhyay D. // Int. J. Hydrogen Energy. 2015. V. 40. P. 12727. https://doi.org/10.1016/j.ijhydene.2015.07.122
Trivedi R., Bandyopadhyay D. // Int. J. Hydrogen Energy. 2016. V. 41. P. 20113. https://doi.org/10.1016/j.ijhydene.2016.09.007
Maltsev A.P., Charkin O.P. // Russ. J. Inorg. Chem. 2020. V. 65. P. 185. https://doi.org/10.1134/S0036023620020114
Maltsev A.P., Charkin O.P. // Russ. J. Inorg. Chem. 2020. V. 65. P. 1204. https://doi.org/10.1134/S0036023620080100
Charkin O.P., Maltsev A.P. // J. Phys. Chem. A. 2021. V. 125. P. 2308. https://doi.org/10.1021/acs.jpca.1c00211
Becke A.D. // J. Phys. Chem. 1993. V. 98. P. 5648. https://doi.org/10.1063/1.464304
Frisch M.J., Trucks G.W., Schlegel H.B. et al. // Gaussian, 09. Revision A.02 Gaussian, Inc., Wallingford CT, 2013.
Charkin O.P., Klimenko N.M., Charkin D.O. // Chem. Phys. 2019. V. 522. P. 112. https://doi.org/10.1016/j.chemphys.2019.02.007
Дополнительные материалы
- скачать ESM.doc
- Figure S1. - Figure S4.
Инструменты
Журнал неорганической химии