

Журнал неорганической химии, 2021, T. 66, № 12, стр. 1647-1665
Ортофосфаты церия(IV) (обзор)
Т. О. Козлова a, А. Е. Баранчиков a, В. К. Иванов a, *
a Институт общей и неорганической химии им. Н.С. Курнакова РАН
119991 Москва, Ленинский пр-т, 31, Россия
* E-mail: van@igic.ras.ru
Поступила в редакцию 14.06.2021
После доработки 05.07.2021
Принята к публикации 06.07.2021
Аннотация
Обзор посвящен анализу сведений о составе, структуре и свойствах аморфных и кристаллических ортофосфатов церия(IV), опубликованных в период с 1882 по 2020 гг. Обзор включает в себя информацию о комплексообразовании Ce(IV) в водных растворах, условиях формирования и характеристиках аморфных неорганических и органо-неорганических гидроортофосфатов церия(IV), структурном разнообразии кристаллических ортофосфатов церия(IV). Впервые проведено сопоставление строения и условий формирования ближайших химических аналогов ортофосфатов церия(IV) ‒ ортофосфатов металлов IV группы и актинидов (тория и урана), на основании которого проанализированы возможности получения еще не описанных изоструктурных соединений церия(IV).
ВВЕДЕНИЕ
Редкоземельные элементы (РЗЭ) представляют собой группу из 17 элементов, включающую лантаниды La–Lu, Sc и Y. Ввиду схожего электронного строения и близости ионных радиусов химические свойства РЗЭ во многом совпадают. Все РЗЭ могут существовать в степени окисления +3, однако для некоторых элементов также характерны степени окисления +2 (Sm, Eu, Yb) и +4 (Ce, Pr, Tb) [1–5]. Редкоземельные элементы в степени окисления +3 легко образуют соединения с ортофосфат-анионами; в зависимости от радиуса катиона эти соединения могут кристаллизоваться в четырех структурных типах: монацит или рабдофан (характерны для легких лантанидов), ксенотим или черчит (характерны для тяжелых лантанидов) [6]. Примечательно, что одним из основных промышленных источников РЗЭ являются именно фосфатсодержащие соединения ‒ монацитовые руды (Ln,Th)PO4 и ксенотим YPO4 [7, 8]. Ортофосфаты РЗЭ обладают рядом практически значимых свойств, в том числе высокой термической стабильностью (температура плавления ~2300°С), низкой теплопроводностью, высокими показателями преломления, высоким квантовым выходом люминесценции [9]. В связи с этим они находят применение в качестве люминофоров, сенсоров, катализаторов [10–14], матриц для захоронения радиоактивных ядерных отходов, а также термостойких керамических материалов [15]. Среди ортофосфатов РЗЭ особое положение занимают соединения церия, включая монацит (CePO4) и рабдофан (CePO4 ⋅ ⋅ xH2O), которые встречаются в природе в виде минералов, а также могут быть получены синтетическим путем [16–21]. Сведения об особенностях их структуры [22–25] и практически значимых свойствах [26–29] подробно освещены в многочисленных научных публикациях.
Помимо ортофосфатов трехвалентного церия известны и ортофосфаты церия(IV) [30]. Несмотря на то, что электронная конфигурация Ce4+ ([Xe]) является более устойчивой по сравнению с Ce3+ ([Xe]4f1), в природе ортофосфаты церия(IV) в составе минералов не встречаются. Узкий диапазон условий образования ортофосфатов церия(IV) обусловлен склонностью Ce(IV) к восстановлению до Ce(III) [31], а также высокой скоростью гидролиза Ce(IV) в водных средах с образованием чрезвычайно стабильного диоксида церия [32].
В настоящем обзоре впервые систематически рассмотрены сведения о составе, структуре и свойствах ортофосфатов церия(IV), охватывающие более чем вековой период развития химии этих соединений. Основные разделы обзора посвящены описанию представителей семейства ортофосфатов церия(IV), известных к настоящему времени (аморфных и кристаллических гидроортофосфатов, кристаллических двойных и смешанных ортофосфатов церия(IV)).
КОМПЛЕКСООБРАЗОВАНИЕ ЦЕРИЯ(IV) В ОРТОФОСФАТНЫХ ВОДНЫХ РАСТВОРАХ
Синтез ортофосфатов церия(IV) обычно осуществляют методами мягкой химии при взаимодействии водных растворов солей церия(IV) с ортофосфорной кислотой или ее солями. Ортофосфорная кислота диссоциирует по трем ступеням (K1 = 10–2.13, K2 = 10–7.21, K3 = 10–12.36), и в зависимости от рН среды в ее растворах преимущественно присутствуют ионы ${{{\text{H}}}_{2}}{\text{PO}}_{4}^{ - }$, ${\text{HPO}}_{4}^{{2 - }}$, ${\text{PO}}_{4}^{{3 - }}$ [33, 34]. В сильноконцентрированных растворах H3PO4 могут происходить процессы ее автопротолиза и поликонденсации [35, 36]. Среди четырехвалентных РЗЭ только соединения церия стабильны в водных растворах [31], однако данные о составе комплексов церия(IV) в фосфатных растворах до сих пор практически отсутствуют. Лебедев и Куляко [37] определяли состав и константы устойчивости фосфатных комплексов Сe(IV) в 3.25–15.00 М растворах H3PO4 измерением окислительного потенциала пары Ce(IV)–Ce(III). Было показано, что в таких условиях наиболее вероятно существование комплекса ${\text{Ce}}({{{\text{H}}}_{2}}{\text{P}}{{{\text{O}}}_{4}})_{3}^{ + }$, ${\text{lg}}\beta _{i}^{0}$ = = 13.08 ± 0.03. В свою очередь, Кониг и Мейн [38] предполагали существование ионов (Ce–O–Ce)6+ в ортофосфатных растворах Ce(IV).
Необходимо отметить, что сведения о составе комплексов Ce(III) в растворах H3PO4 также крайне фрагментарны. Бирн и др. [39] отмечали, что комплексообразование Ce(III) в ортофосфорной кислоте при низких значениях рН нужно рассматривать с учетом образования дигидро- и гидрофосфатных комплексов. Чиркст и Черемисина [40] показали, что состав растворов Ce(III) в H3PO4 существенно зависит от температуры и концентрации кислоты. Так, раствор, полученный растворением CePO4 ⋅ 0.5H2O в 4.5 М H3PO4 при температуре 25°С, содержит ионы Ce(H2PO4)2+ и ${\text{Ce}}({{{\text{H}}}_{2}}{\text{P}}{{{\text{O}}}_{4}})_{2}^{ + }$ (62 и 36% соответственно). При температуре 80°С и концентрации ортофосфорной кислоты 1.2 М это соотношение меняется до 55 и 44% соответственно.
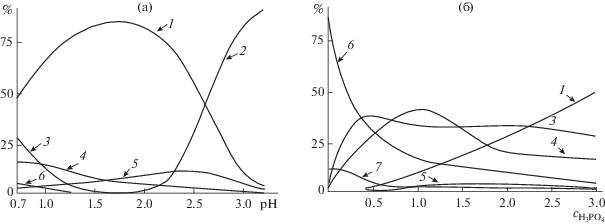
Данные о комплексах церия(IV) в растворах ортофосфорной кислоты полезно проанализировать, принимая во внимание сведения о комплексах Th(IV) ‒ химического аналога Ce(IV) [41]. Авторами [42] был определен ионный состав фосфатных растворов тория(IV) в зависимости от рН и концентрации H3PO4 (рис. 1). Видно, что дигидроортофосфаты, в том числе гидроксодигидроортофосфаты, являются преобладающими ионными формами тория(IV) при низких значениях рН в разбавленных растворах ортофосфорной кислоты. С большой долей вероятности можно предположить, что аналогичные комплексы присутствуют и в фосфатных растворах церия(IV), получаемых в схожих условиях.
Рис. 1.
Мольная доля комплексов фосфатов тория(IV) в зависимости от рН (а) и концентрации ортофосфорной кислоты (б). 1 – Th(H2PO4)4, 2 – Th(H2PO4)3(HPO4$)_{2}^{{3 - }}$ или Th(OH)2(H2PO4$)_{5}^{{3 - }}$, 3 – Th(H2PO4$)_{2}^{{2 + }}$, 4 – ThOH(H2PO4$)_{2}^{ + }$, 5 – ThOH(H2PO4)3, 6 – ThOH(H2PO4)2+, 7 – Th(OH)2(H2PO4)+ [42].
Недостаток информации о составе комплексов церия(IV) в фосфатных растворах затрудняет описание составов формирующихся из них твердофазных соединений, особенно аморфных. Аморфным ортофосфатам церия(IV), в том числе получаемым в виде гелей, посвящено достаточно большое число публикаций, выходивших в свет начиная с конца XIX века, однако большинство этих работ сфокусировано на практическом применении этих материалов, но не на их составе и структуре.
АМОРФНЫЕ ОРТОФОСФАТЫ ЦЕРИЯ(IV)
Впервые гелеобразные осадки аморфных ортофосфатов церия(IV) были получены Хартли [43] в 1882 г. смешением растворов фосфата натрия и нитрата (либо сульфата) церия(IV). Одному из продуктов синтеза была приписана формула ${\text{Ce}}_{4}^{{{\text{IV}}}}{{{\text{H}}}_{2}}$(PO4)6 ⋅ 25H2O. В 70-е гг. XX века Альберти и др. [44] было установлено, что аморфный продукт взаимодействия раствора сульфата церия(IV) с раствором ортофосфорной кислоты при соотношении P/Ce = 2 характеризуется волокнистой микроструктурой. Полученному соединению был приписан состав Ce(HPO4)2 ⋅ H2O. Альберти и др. полагали это соединение церия(IV) кристаллическим, однако, согласно приведенным ими данным рентгенофазового анализа [44], продукт скорее следует назвать аморфным или слабозакристаллизованным, что подтверждают более поздние работы других авторов (например [45, 46]), воспроизводивших методику Альберти и др. [44]. Дель Рей-Буено и др. [45] исследовали термическое поведение волокнистого Ce(HPO4)2 ⋅ H2O и подтвердили достоверность приписанного ему в [44] состава. На основании результатов термического анализа они сделали вывод о том, что в интервале температур до 200°С из Ce(HPO4)2 ⋅ H2O удаляется вода, а при температурах свыше 500°С происходит изменение степени окисления церия +4 на +3 и выделение кислорода. Отметим, что термолиз волокнистого гидроортофосфата тория(IV) Th(HPO4)2 ⋅ 2.5H2O, получаемого смешением раствора нитрата тория с раствором ортофосфорной кислоты [47], происходит без выделения кислорода с образованием при температурах свыше 400°С ThP2O7, что было продемонстрировано Перез-Хименез и др. [48]. Барба и др. [49] исследовали протонную проводимость Ce(HPO4)2 ⋅ H2O, полученного по методике Альберти и др., и показали, что она определяется степенью гидратированности поверхности. Каскиола и др. [50] выявили, что при 20°С и относительной влажности 90% проводимость волокнистого Ce(HPO4)2 ⋅ H2O составляет от 3 × 10–5 до 10–4 См/см и уменьшается до 5 × × 10–7 См/см при относительной влажности 11%.
Волокнистую микроструктуру аморфных ортофосфатов церия(IV), получаемых в том числе в виде гелей, наблюдали и другие исследователи, однако механизм ее формирования достоверно не установлен и до недавнего времени вообще не изучался. Отметим, что сведения об одномерных неорганических материалах, получаемых прямым безтемплатным золь-гель методом, вообще крайне скудны. Известные к настоящему времени примеры, помимо аморфных ортофосфатов церия(IV) и тория(IV), включают в себя гели пентаоксида ванадия [51] и AgVO3 [52]. В 2018 г. Ёровым и др. [53] было высказано предположение, что гели ортофосфатов церия(IV), синтезированные смешением церийфосфатных растворов с водой или некоторыми апротонными растворителями, образуются по полимеризационному механизму [54]. Козловой и др. [55] был выполнен более детальный анализ механизма формирования гелей ортофосфатов церия(IV). При добавлении воды к церийфосфатному раствору, вероятно, происходит диссоциация групп P–O–H комплексов ортофосфатов церия(IV), присутствующих в растворе, с образованием H3O+ и анионных комплексов дигидроортофосфатов церия(IV). Последние, будучи нуклеофильными, могут реагировать с другими аналогичными комплексами ортофосфатов церия(IV), образуя мостики Ce‒[PO4]–Ce, что является первым этапом образования церийфосфатного каркаса. Отметим, что при рассмотрении данного механизма стоит также учитывать склонность Ce(IV) к гидролизу [56] и поэтому принимать во внимание возможность частичного гидролиза связей Ce–O–P с последующим образованием связей Сe–O–H и Ce–O–Ce, что наблюдается для ортофосфатов некоторых других четырехвалентных металлов [57, 58].
При этом pH реакционной среды может влиять на скорость гидролиза связей Ce–O–P и диссоциации комплексов ортофосфатов церия(IV) и, как следствие, определять скорость образования геля. Для подтверждения данной гипотезы Козловой и др. [55] были изучены условия формирования монолитных гелей ортофосфатов церия(IV) в зависимости от состава и объема гелирующего агента (вода, водные растворы H3PO4, HNO3 или H2SO4), добавляемого к церийфосфатному раствору. Использование растворов минеральных кислот вместо воды замедляло процесс гелеобразования, что согласуется с наблюдениями Пармара и др. [59], сделанными при исследовании гелеобразования ортофосфатов тория(IV). Было показано, что при использовании 3 М раствора ортофосфорной кислоты могут быть получены монолитные гели, в которых на один атом церия в церийфосфатном каркасе приходится около 20 000 молекул воды. Ёровым и др. [53], а также Козловой и др. [55] из монолитных гелей ортофосфатов церия(IV) методом сверхкритической сушки были получены аэрогели, обладающие рекордной для класса неорганических неуглеродных аэрогелей геометрической плотностью 1 мг/см3. Методом малоуглового рассеяния нейтронного излучения (МУРН) на мезоскопическом уровне была проанализирована структура полученных аэрогелей. Было показано, что скорость формирования гелей влияет на размер элементов структуры аэрогелей ортофосфатов церия(IV) – чем выше скорость конденсации, тем меньший размер имеют рассеивающие неоднородности.
Метод МУРН имеет ограничения для исследований гидратированных гелей из-за некогерентного рассеивания на ядрах водорода. Амарантовым и др. [60] впервые был использован метод малоуглового рассеяния рентгеновского излучения для изучения структуры гелей ортофосфатов церия(IV), полученных из растворов с различными концентрациями церия и не подвергавшихся сушке. Было показано, что увеличение концентрации церия в исходных растворах приводит к уменьшению расстояния между рассеивающими неоднородностями в получаемых гелях ортофосфатов церия(IV), а также к увеличению степени агрегации и изменению структуры гелей от объемного к поверхностному фракталу.
Большинство работ, связанных с исследованиями аморфных ортофосфатов церия(IV), посвящены анализу их возможных практических приложений, связанных прежде всего с ионообменными процессами. Первыми анализ ионообменных свойств аморфных ортофосфатов церия(IV) выполнили в середине XX века Виссерс и Рокко [61]. Ларсен и Цилли [62] синтезировали гель ортофосфата церия(IV), которому формально был приписан состав Ce3(OH)8(H2PO4)4. Было показано, что полученное соединение способно к обмену ионов H+ на Li+, Na+ и K+, при этом рассчитанная стандартная энтальпия реакций составила 0.87 ± 0.13, 0.75 ± 0.15 и ‒0.15 ± 0.03 ккал/моль соответственно. Альберти и др. [44, 63] исследовали ионообменные характеристики волокнистого Ce(HPO4)2 · H2O. Измеренная ионообменная емкость гидроортофосфата церия(IV) составила 5 мэкв/г. Хаяши и др. [64] показали, что селективность ионного обмена на Ce(HPO4)2 ⋅ H2O возрастает в следующей последовательности: Na+ $ \ll $ K+ < < Rb+ < Cs+, Mg2+ < Ca2+ < Sr2+ < Ba2+. Альберти и др. [65] на основании результатов экспериментов по изучению сорбции на Ce(HPO4)2 · H2O предположили, что в структуре данного ионообменного материала присутствует около 25‒30% позиций, занятых протонами, способными к обмену. Значение ионообменной емкости гидроортофосфата церия(IV), полученного по методике Альберти и др., не уступает и в ряде случаев превосходит соответствующие значения для известных неорганических ионообменных материалов [66]. Для практического применения, в том числе для целей хроматографии, Альберти и др. [67] на основе волокнистого гидроортофосфата церия(IV) получили ионообменную бумагу, обладающую высокой механической прочностью и химической стабильностью, а также высокой селективностью по отношению к катионам K+, Cs+, Rb+, Ag+, Fe2+ и Pb2+. Альберти и Костантино [47] предложили использовать в аналогичных целях волокнистый гидроортофосфат тория(IV), имеющий, однако, более низкую ионобменную емкость по сравнению с цериевым аналогом ‒ 3.7 мэкв/г. Анил и Чаудри [68] синтезировали гидроортофосфаты тория(IV) по методикам, близким к методике Альберти и Костантино [47], однако имеющие повышенные величины ионообменной емкости ‒ до 4.54 мэкв/г.
Хаяши и др. [46] разработали эффективный метод извлечения катионов стронция из радиоактивных водных растворов волокнистым Ce(HPO4)2 · H2O, включающий в себя гидротермальную обработку реакционной смеси при температурах до 250°С. Было показано, что в ходе гидротермальной обработки меняется структура исходного гидроортофосфата церия(IV) и формируется продукт предполагаемого состава (${\text{Ce}}_{{2 - 2y}}^{{3 + }}{\text{Ce}}_{y}^{{4 + }}{\text{Sr}}_{y}^{{2 + }}$)(PO4)2, имеющий структуру, близкую структуре монацита.
Романчук и др. [69] было продемонстрировано, что сорбент на основе волокнистого гидроортофосфата церия(IV) способен формироваться при непосредственном добавлении церийфосфатного раствора к жидким радиоактивным отходам. Наибольшая степень извлечения радионуклидов из реакционных сред, в том числе из модельных растворов реальных радиоактивных отходов, была зафиксирована для Th(IV). Было показано [69], что отжиг получаемого сорбента, содержащего торий, при температуре 1200°С приводит к кристаллизации замещенного ортофосфата церия(III) со структурой монацита ‒ стабильной матрицы для иммобилизации радиоактивных отходов [15, 22, 26, 27].
В 2000-е гг. был издан ряд работ, посвященных исследованию гибридных ионообменных материалов на основе волокнистых ортофосфатов церия(IV) и органической составляющей. Сочетание органической и неорганической компонент способствует достижению высокой воспроизводимости ионообменных свойств материалов на основе ортофосфатов церия(IV), а также обеспечивает их долговременную химическую и механическую стабильность [70–72]. Варшней и др. [73] показали, что величина адсорбции щелочноземельных и тяжелых металлов на волокнистом ортофосфате церия(IV) зависит от присутствия и типа поверхностно-активных веществ (ПАВ) в растворе. Так, мицеллы анионных ПАВ уменьшали адсорбцию тяжелых металлов (кроме Hg(II)) на ортофосфате церия(IV) при неизменной величине адсорбции щелочноземельных металлов. Напротив, мицеллы катионных и неионогенных ПАВ увеличивали адсорбцию как щелочноземельных, так и тяжелых металлов. В дальнейшем Варшней, Икбал и Сома синтезировали органо-неорганические композиты на основе волокнистого ортофосфата церия(IV), в которых в качестве органической составляющей выступали поверхностно-активные вещества, а именно: додецилсульфат натрия [74], Тритон Х-100 [75], N-додецилпиридиний хлорид [76], цетилпиридиний хлорид [77], бис(2-этилгексил)сульфосукцинат натрия [78], додецилбензолсульфонат натрия [79]. Для получения композитов раствор 0.05 М сульфата церия в объемном соотношении 1 : 2 при нагревании прикапывали к 6 М раствору ортофосфорной кислоты и соответствующего ПАВ. Полученную суспензию выдерживали в течение 3.5 ч при температуре 60°С, затем промывали до pH ~ 4 и высушивали. В H+-форму полученный материал переводили обработкой в 1 M HNO3 в течение 24 ч [80]. Катионообменная емкость волокнистого ортофосфата церия(IV) без добавления ПАВ составила 1.3 мэкв/г по отношению к Na+; использование ПАВ позволило увеличить емкость до 2.12–3.15 мэкв/г в зависимости от концентрации и типа ПАВ. Повышение ионообменной емкости гибридных материалов авторы связали с увеличением межслоевого пространства в структуре ортофосфата церия(IV) вследствие интеркаляции ПАВ, а также с уменьшением поверхностного натяжения между твердой и жидкой фазами. Было установлено, что полученные органо-неорганические материалы перспективны для применения в областях, связанных с очисткой воды, поскольку обладают селективностью по отношению к ионам тяжелых металлов, включая Pb(II) и Hg(II) [74, 79]. Эл-Азони и др. [80] показали, что композит ортофосфат церия(IV)‒Тритон Х-100 может быть использован для эффективного разделения Cr(III) и Cr(VI): в разбавленной HCl Cr(III) прочно связывается с таким композитом, в то время как Cr(VI) остается в растворе. Ионообменная емкость материала составила 2.1 мэкв/г по отношению к Cr(III).
Аналогичные исследования были проведены с использованием других органических составляющих композитов, в том числе полимерных соединений, в частности, были получены композиты на основе аморфного ортофосфата церия(IV) и акрилонитрила (2.86 мэкв/г) [81], акриламида (2.6 мэкв/г) [82], пектина (1.5 мэкв/г) [83]. Для сравнения величины ионообменной емкости для аналогичных композитов ортофосфат тория(IV)–акриламид и ортофосфат тория(IV)–пектин составили 2.0 [76] и 1.57 мэкв/г [83] соответственно.
Метуолли и др. с использованием схожих синтетических подходов получили композиты ортофосфат церия(IV)–полиакриламид [84] и ортофосфат церия(IV)–полиакрилонитрил [85]. Было показано, что такие материалы перспективны для извлечения и разделения радиоактивных изотопов 60Co, 134Cs, 152 + 154Eu.
Шакшуки и др. [86] для получения органо-неорганического церийфосфатного композита использовали несколько иной подход – предварительно синтезированный волокнистый ортофосфат церия(IV) состава Ce(HPO4)2 ⋅ 2.9H2O (ионообменная емкость 5.21 мэкв/г) вымачивали в этанольном растворе бензимидазола в течение 3 сут. В результате полимеризации бензимидазола происходило формирование целевого продукта. При этом полимеризация, по мнению авторов, была обусловлена in situ окислением мономера за счет частичного восстановления ионов Ce(IV), присутствующих в неорганической матрице.
КРИСТАЛЛИЧЕСКИЕ ОРТОФОСФАТЫ ЦЕРИЯ(IV): ГИДРООРТОФОСФАТЫ
Интерес к кристаллическим ортофосфатам церия(IV), согласно имеющимся литературным данным, возник примерно в то же время, что и к аморфным. В частности, Херман и Клирфилд [87] добавлением ортофосфорной кислоты к раствору (NH4)2Ce(NO3)6 при нагревании получили шесть кристаллических ортофосфатов церия(IV) различного брутто-состава за счет варьирования условий синтеза. Достоверность определения состава этих соединений в более поздних работах ставилась под сомнение [88, 89]. Херман и Клирфилд [87] продемонстрировали, что полученные соединения состава Ce(OH)0.45(PO4)0.45(HPO4)1.1 ⋅ 0.33H2O, Ce(OH)0.375(PO4)0.375[(NH4)0.09H1.16(PO4)1.25] ⋅ 0.25H2O и Ce(NH4PO4)0.44(HPO4)1.56 проявляют ионообменные свойства в щелочной среде и характеризуются значениями ионообменной емкости около 3‒4 мэкв/г.
Цухако и др. [90] из суспензии CeO2 в 85%-ной H3PO4 в гидротермальных условиях получили неописанный ранее кристаллический ортофосфат церия(IV), которому на основании данных ИК-спектроскопии, рентенофазового и термогравиметрического анализа приписали состав Ce(HPO4)2 ⋅ 2H2O. Было показано, что данное соединение имеет слоистую структуру с межслоевым расстоянием 18.0 Å, которое обратимо изменяется в зависимости от влажности атмосферы. Авторы [90] также продемонстрировали, что Ce(HPO4)2 ⋅ 2H2O является эффективным сорбентом ионов аммония.
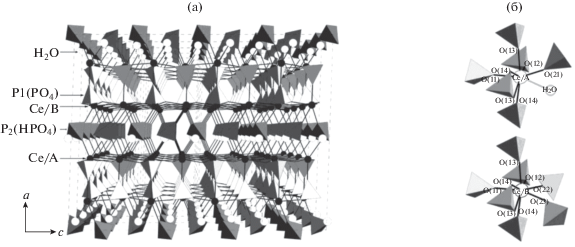
В 2005 г. Назарали и др. [91] было впервые достоверно охарактеризовано соединение состава Ce(PO4)(HPO4)0.5(H2O)0.5 и решена его структура11. Синтез осуществляли гидротермальной обработкой нанокристаллического диоксида церия в разбавленной ортофосфорной кислоте. Было показано, что структура нового гидроортофосфата церия(IV) является слоистой и представлена двойными слоями [Ce(PO4)+]n. Каждый атом церия связан с четырьмя ортофосфатными группами, образующими одну сторону слоя, и с одной ортофосфатной группой на противоположной стороне этого слоя. Между слоями находятся гидроортофосфатные группы ${\text{HPO}}_{4}^{{2 - }}$ и молекулы воды. Особенностью структуры этого соединения являются два варианта координации атомов церия, которые помимо ортофосфатных групп сочленены либо с гидроортофосфатной группой и молекулой воды, либо с двумя гидроортофосфатными группами (рис. 2). При этом последние прочно связывают слои вместе, что затрудняет интеркаляцию ряда соединений, например аминов, в межслоевое пространство.
Таблица 1.
Состав и структурные параметры ортофосфатов церия(IV) и их изоструктурных аналогов
| № | Состав | Пр. гр. | а, Å | b, Å | c, Å | α, β, γ, град |
|---|---|---|---|---|---|---|
| 1 | Сe(PO4)1.5(H2O)(H3O)0.5(H2O)0.5 [89] | С2/c | 15.7058(17) | 9.6261(9) | 10.1632(4) | β = 121.623(7) |
| 2 | Ce(PO4)(HPO4)0.5(H2O)0.5 [91] | С2/с | 21.0142(3) | 6.55082(7) | 6.94382(6) | β = 91.983(1) |
| 3 | U2(PO4)2HPO4 · H2O [96] | С2/с | 21.148(7) | 6.611(2) | 6.990(3) | β = 91.67(3) |
| 4 | Th2(PO4)2HPO4 · H2O [99] | н.у.* | 21.368(2) | 6.695(1) | 7.023(1) | α = β = γ = 90 |
| 5 | (NH4)2Ce(PO4)2 · H2O [129] | Imma | 6.88940(9) | 6.88860(9) | 17.723(2) | α = β = γ = 90 |
| 6 | (NH4)2Th(PO4)2 · H2O [116] | I41/amd | 7.0192(4) | 7.0192(4) | 17.9403(8) | α = β = γ = 90 |
| 7 | NH4Ce2(PO4)3 [115] | С2/с | 17.4719(4) | 6.7693(1) | 7.9929(1) | β = 102.873(1) |
| 8 | NH4Th2(PO4)3 [116] | С2/с | 17.7238(5) | 6.90676(12) | 8.15603(32) | β = 102.1047(26) |
| 9 | KTh2(PO4)3 [117] | С2/с | 17.57 | 6.863 | 8.138 | β = 101.46 |
| 10 | Na10Ce2(PO4)6 (Na5Ce(PO4)3) [132] | P212121 | 6.9375(14) | 16.215(3) | 18.765(4) | α = β = γ = 90 |
| 11 | K2Ce(PO4)2 [134] | P21/n | 9.1060(4) | 10.8160(5) | 7.6263(4) | β = 111.155(2) |
| 12 | K2Ce(PO4)2 (t = 610°C) [136] | Imma | 6.8363(3) | 6.8369(3) | 17.5090(1) | α = β = γ = 90 |
| 13 | K2Ce(PO4)2 (t = 610°C) [136] | I41/amd | 6.83662(3) | 6.83662(3) | 17.5091(1) | α = β = γ = 90 |
| 14 | K2Th(PO4)2 [138] | P21/n | 9.1956(4) | 10.9562(3) | 7.7114(3) | β = 111.437(4) |
| 15 | K2Th(PO4)2 (t = 850°C) [137] | I41/amd | 6.9593(2) | 6.9593(2) | 17.6699(9) | α = β = γ = 90 |
| 16 | K4CeZr(PO4)4 [139] | I41/amd | 6.7039(9) | 6.7039(9) | 17.065(3) | α = β = γ = 90 |
| 17 | Ce(OH)PO4 [145] | Cmce | 6.9691(3) | 9.0655(4) | 12.2214(4) | α = β = γ = 90 |
| 18 | Th(OH)PO4 [148] | Cmca | 7.1393(2) | 9.2641(2) | 12.5262(4) | α = β = γ = 90 |
| 19 | U(OH)PO4 [148] | Cmca | 7.0100(2) | 9.1200(2) | 12.3665(3) | α = β = γ = 90 |
| 20 | Ce2O(PO4)2 [145] | Cmce | 7.0220(4) | 8.9894(5) | 12.544(1) | α = β = γ = 90 |
| 21 | Th2O(PO4)2 [148] | Cmca | 7.1691(3) | 9.2388(4) | 12.8204(7) | α = β = γ = 90 |
| 22 | U2O(PO4)2 [151] | Cmca | 7.088 | 9.037 | 12.702 | α = β = γ = 90 |
| 23 | Np2O(PO4)2 [151] | Cmca | 7.038 | 9.015 | 12.603 | α = β = γ = 90 |
| 24 | β-Zr2O(PO4)2 [153] | Cmca | 6.624(1) | 8.637(2) | 11.872(2) | α = β = γ = 90 |
| 25 | (NH4)[CeF2(PO4)] [159] | P21/m | 6.660(2) | 5.875(2) | 7.177(3) | β = 114.31(2) |
| 26 | (NH4)[CeF2(AsO4)] [168] | P21/m | 6.7758(2) | 6.0181(2) | 7.1315(2) | β = 113.832(2) |
| 27 | [(CH2)2(NH3)2]0.5[CeF3(HPO4)] [160] | P1 | 6.248(2) | 7.079(2) | 8.794(3) | α = 103.92(2) β = 100.84(2) γ = 110.28(2) |
Назарали и др. [91] установили, что ранее авторами [92] гидротермальной обработкой волокнистого аморфного ортофосфата церия(IV) в среде ортофосфорной кислоты был синтезирован аналогичный слоистый гидроортофосфат церия(IV), однако ему был ошибочно приписан состав Ce(HPO4)(PO4)0.5(OH)0.5. Хаяши и др. [92] показали, что продукт гидротермального синтеза имеет ионообменную емкость, в несколько раз меньшую, чем емкость исходного аморфного соединения (1 и 4.5 мэкв/г соответственно), что согласуется с наблюдениями Назарали и др. [91].
Шекунова и др. [93] исследовали влияние содержания ортофосфорной кислоты в реакционной среде на состав продуктов гидротермальной обработки аморфных гелей ортофосфата церия(IV). В частности, было обнаружено, что использование 11%-ного раствора H3PO4 приводит к кристаллизации Ce(PO4)(HPO4)0.5(H2O)0.5, что согласуется с результатами Хаяши и др. [92], а гидротермальная обработка в воде – к формированию Ce(PO4) ⋅ xH2O.
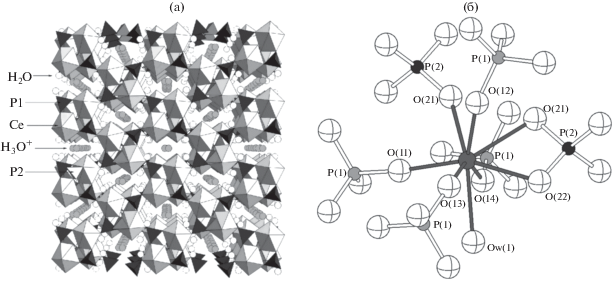
Для получения Ce(PO4)(HPO4)0.5(H2O)0.5 Назарали и др. [88, 89] успешно апробировали и другой синтетический подход, который заключался в гидротермальной обработке раствора (NH4)2Ce(NO3)6 в смеси разбавленных азотной и ортофосфорной кислот при 95°С в течение 5 сут. Оказалось, что в этом случае кристаллизация Ce(PO4)(HPO4)0.5(H2O)0.5 проходит через стадию формирования стабильного однофазного продукта состава Ce(PO4)1.5(H2O)(H3O)0.5(H2O)0.5, структура которого была решена методом монокристальной рентгеновской дифракции. Особенностью данного соединения является наличие туннелей, расположенных вдоль оси с и содержащих молекулы воды и катионы H3O+ (рис. 3). Трансформацию Ce(PO4)1.5(H2O)(H3O)0.5(H2O)0.5 в Ce(PO4)(HPO4)0.5(H2O)0.5 в ходе старения реакционной смеси авторы объяснили процессами растворения-рекристаллизации. Назарали и др. отметили, что полученные ими структурные данные совпадают с результатами работы [94], авторы которой, однако, приписали продукту синтеза неверный состав ‒ Ce(OH)1.62(NH4HPO4)0.35(H2PO4)0.68(PO4)0.45 · 0.6H2O. Наличие туннелей и присутствие в них H3O+ обусловливает способность Ce(PO4)1.5(H2O) (H3O)0.5(H2O)0.5 к ионному обмену, что согласуется с выводами Хермана и Клирфилда [94]. Назарали и др. [89] показали, что селективность ионного обмена уменьшается в ряду Li+ > Na+ = K+ > Rb+ > Cs+ и связали это со стерическими факторами.
В работе [95] были изучены сорбционные свойства Ce(PO4)(HPO4)0.5(H2O)0.5 по отношению к радионуклидам 239Np, 241Am, 233, 232U, 234Th, 90Sr, 137Cs в зависимости от рН среды. Было установлено, что сорбция актинидов происходит только на поверхности Ce(PO4)(HPO4)0.5(H2O)0.5 без их встраивания в структуру соединения.
Идентичное гидроортофосфату церия(IV) Ce(PO4)(HPO4)0.5(H2O)0.5 соединение номинального состава Ce2(PO4)2HPO4 ⋅ H2O было получено Бранделом и др. [96] при взаимодействии смеси 1 М раствора (NH4)2Ce(NO3)6 с 5 М раствором ортофосфорной кислоты и 2 М раствором азотной кислоты в гидротермальных условиях при 150°С в течение минимум одной недели.
Сато и др. [97, 98] показали, что Ce2(PO4)2HPO4 ⋅ H2O является полупроводником с шириной запрещенной зоны ~2.7 эВ и имеет низкую активность в реакциях каталитического окисления органических соединений. При этом коэффициент поглощения УФ-излучения у гидроортофосфата церия(IV) оказался выше по сравнению с CePO4 и CeP2O7, что в совокупности с другими перечисленными факторами делает Ce2(PO4)2HPO4 ⋅ H2O перспективным компонентом солнцезащитной косметики.
Для Ce2(PO4)2HPO4 ⋅ H2O известны изоструктурные торий- и урансодержащие аналоги. Дашо и др. [99] получали Th2(PO4)2HPO4 ⋅ H2O двумя способами: гель, сформированный при смешении растворов нитрата или хлорида тория и ортофосфорной кислоты в мольном соотношении Th : PO4 = 2 : 3, нагревали в закрытой емкости при 150–160°С в течение нескольких часов либо выдерживали при 160°C в автоклаве в течение месяца. Примечательно, что структура полученного продукта была описана в ромбической сингонии, отличной от сингонии Ce2(PO4)2HPO4 ⋅ H2O, однако Брандел и др. [96] показали, что разница в описании может быть обусловлена незначительным искажением элементарной ячейки Ce2(PO4)2HPO4 ⋅ ⋅ H2O вследствие различия в ионных радиусах тория и церия ($^{{{\text{VII}}}}{{r}_{{{\text{T}}{{{\text{h}}}^{{{\text{4 + }}}}}}}}$ = 0.100 нм и $^{{{\text{VII}}}}{{r}_{{{\text{C}}{{{\text{e}}}^{{{\text{4 + }}}}}}}}$ = 0.092 нм).
Сальвадо и др. [100] сообщили о получении Th2(PO4)2HPO4 ⋅ H2O гидротермальной обработкой смеси H3PO3 и Th(NO3)4 ⋅ 5H2O, однако их результаты решения структуры продукта синтеза не совпали с кристаллографическими данными, представленными Дашо и др. [99] или Бранделом и др. [96] для соединения такого же состава.
Бранделом и др. [96] был синтезирован изоструктурный аналог торий- и церийсодержащих фаз состава U2(PO4)2HPO4 ⋅ H2O гидротермальной обработкой UCl4 или UO2 в растворе ортофосфорной кислоты, а также термогидролизом U(HPO4)2 ⋅ nH2O. Полученная фаза была описана в моноклинной сингонии, как и Ce2(PO4)2HPO4 ⋅ ⋅ H2O ($^{{{\text{VII}}}}{{r}_{{{{{\text{U}}}^{{{\text{4 + }}}}}}}}$ = 0.095 нм и $^{{{\text{VII}}}}{{r}_{{{\text{C}}{{{\text{e}}}^{{{\text{4 + }}}}}}}}$ = 0.092 нм).
Дашо и др. [101] были предприняты попытки получения твердых растворов ${\text{T}}{{{\text{h}}}_{{2--x/2}}}{\text{An}}_{{{x \mathord{\left/ {\vphantom {x 2}} \right. \kern-0em} 2}}}^{{{\text{IV}}}}$(PO4)2(HPO4) · ⋅ H2O (An = U, Np, Pu) гидротермальной обработкой гелеобразных осадков, полученных при смешении кислых растворов актинидов с ортофосфорной кислотой. Данный подход оказался результативным при использовании мольных отношений An/Th ≤ 1.
Среди возможных структурных аналогов гидроортофосфатов церия(IV) достоверно установлены состав и структура некоторых соединений тория и металлов 4 группы Периодической системы Д.И. Менделеева. В отличие от Ce(HPO4)2 ⋅ ⋅ xH2O, структурные данные для которого отсутствуют, кристаллические гидроортофосфаты МIV(HPO4)2 ⋅ хH2O (M = Ti, Hf, Zr) достаточно подробно изучены. В частности, различают α- и γ-модификации при х = 1 и 2 соответственно. Определение структур соединений ряда МIV(HPO4)2 ⋅ ⋅ H2O (M = Ti, Hf, Zr) показало, что они образованы слоями [МIV(HPO4)2]∞ из слегка искаженных октаэдров МIVO6 и полиэдров HPO4. С увеличением радиуса катиона происходит линейное увеличение параметров и объема элементарной ячейки с сохранением исходной сингонии. В межслоевом пространстве находятся молекулы воды [102, 103], а расстояние между слоями составляет ~7.6 Å. Для соединений состава МIV(HPO4)2 ⋅ 2H2O (или MIV(PO4)(H2PO4) ⋅ 2H2O, M = Ti, Hf, Zr) межслоевое расстояние существенно больше (~12 Å) ввиду присутствия в нем дигидроортофосфатных групп [103, 104].
Сальвадо и др. [105] определили структуру Th(HPO4)2 ⋅ H2O, полученного из смеси Th(NO3)4, CO(NH2)2 и H3PO3 в гидротермальных условиях. Структура Th(HPO4)2 ⋅ H2O представляет собой трехмерный каркас, сформированный полиэдрами ThO6${{{\text{O}}}_{{{{{\text{H}}}_{2}}{\text{O}}}}}$, с небольшими каналами и совершенно отличается от структуры слоистого α-фосфата циркония Zr(HPO4)2 ⋅ H2O.
Таким образом, семейство гидроортофосфатов церия(IV) к настоящему моменту включает всего два соединения ‒ Ce(PO4)(HPO4)0.5(H2O)0.5 и Ce(PO4)1.5(H2O)(H3O)0.5(H2O)0.5. Примечательно, что возможность получения средних ортофосфатов церия(IV), равно как и ортофосфатов актинидов, до сих пор надежно не установлена [106, 107]. При этом количество работ, посвященных ортофосфатам церия(IV), содержащим несколько различных катионов или анионов, с каждым годом увеличивается.
КРИСТАЛЛИЧЕСКИЕ ОРТОФОСФАТЫ ЦЕРИЯ(IV): ДВОЙНЫЕ СОЛИ
Многочисленные работы Орловой и др. [108–111] посвящены получению и изучению соединений общего состава ${{{\text{A}}}^{{\text{I}}}}{\text{Ce}}_{2}^{{{\text{IV}}}}{{({\text{P}}{{{\text{O}}}_{4}})}_{3}}$ (AI = Li–Cs) и E0.5Ce2(PO4)3 (E = Mg‒Ba, Cd). Предложен метод синтеза таких соединений, который заключается в осаждении разбавленной ортофосфорной кислотой из смеси растворов (NH4)2Ce(NO3)6 и соли соответствующего металла с последующим высушиванием и отжигом осадков при температурах 600‒1600°С в течение минимум 1 ч. Согласно данным, представленным в работах [108–111], полученные соединения кристаллизуются в структурном типе монацита и обладают незначительно большим объемом элементарных ячеек по сравнению с CePO4. Орловой и др. [112] также была получена группа изоструктурных двойным ортофосфатам церия(IV) монацитоподобных фаз состава E0.5M2(PO4)3 (E = Mg, Ca, Sr; M = Np, Pu). Достоверность некоторых результатов, представленных Орловой и др., была подвергнута сомнению другими исследователями. В частности, Орловой и др. [113] было получено и описано в структурном типе монацита соединение BаCe(PO4)2, принадлежащее к группе двойных ортофосфатов церия(IV) общего состава ECe(PO4)2 (E = Mg‒Ba, Cd). В работе [114] другими авторами аналогичным методом синтезирован “BаCe(PO4)2” и показано, что, хотя дифрактограмма и соответствует структуре монацита, в продукте также присутствует примесь аморфной фазы, содержащая барий. Кроме того, данные рентгеновской фотоэлектронной спектроскопии указывают на то, что церий в “BaCe(PO4)2” находится в степени окисления +3, что не соответствует сведениям, представленным в работе Орловой и др. [113].
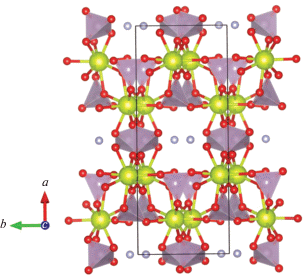
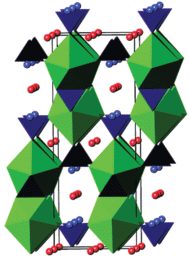
Шекуновой и др. [115] был синтезирован и достоверно структурно исследован двойной ортофосфат церия(IV) состава ${{{\text{A}}}^{{\text{I}}}}{\text{Ce}}_{2}^{{{\text{IV}}}}{{({\text{P}}{{{\text{O}}}_{4}})}_{3}}$, в качестве АI выступал катион аммония. Синтез осуществляли гидротермальной обработкой аморфного геля ортофосфата церия(IV) в среде 0.1‒2 М раствора аммиака. Было показано, что полученный двойной ортофосфат церия(IV)-аммония NH4Ce2(PO4)3 изоструктурен NH4Th2(PO4)3 [116] и KTh2(PO4)3 [117], имеет каркасное строение и кристаллизуется в моноклинной сингонии в пр. гр. C2/c (рис. 4). Отметим, что KTh2(PO4)3 также имеет близкие с NaTh2(PO4)3 параметры элементарной ячейки, однако последнее соединение было описано в пр. гр. Сс [118], что объясняется различным расположением катионов натрия и калия в соответствующих структурах [117]. Отметим, что к структурному типу NaTh2(PO4)3 относится большое семейство изоструктурных соединений, в том числе уранового ряда, например, KU2(PO4)3, NaU2(PO4)3 и LiU2(PO4)3 [119, 120].
Рис. 4.
Структура NH4Ce2(PO4)3. Желтым цветом обозначены атомы церия, светло-голубым ‒ атомы азота, красным – атомы кислорода. Атомы фосфора находятся внутри тетраэдров PO4 (обозначены фиолетовым цветом) [115].
Изоструктурность двойных ортофосфатов тория(IV), содержащих в качестве второго катиона калий или аммоний, объясняется близкими значениями радиусов этих катионов (1.51 [121] и 1.54 Å [122] для КЧ = 8 соответственно). Исходя из этого, для NH4Ce2(PO4)3 можно ожидать существование изоструктурного аналога KCe2(PO4)3, в связи с чем корректность описания Орловой и др. [108] полученного ими соединения номинального состава KCe2(PO4)3 в структурном типе монацита также вызывает сомнения. Шекуновой и др. [115] был выполнен анализ термического разложения NH4Ce2(PO4)3 в сопоставлении с NH4Th2(PO4)3. Согласно данным Сальвадо и др. [116], NH4Th2(PO4)3 характеризуется высокой термической стабильностью вплоть до температур свыше 700°C, после чего в одну стадию формируется β-Th4(PO4)4(P2O7). Напротив, термическое разложение NH4Ce2(PO4)3 носит многостадийный характер и начинается при гораздо более низких температурах. Разница в термическом поведении изоструктурных фаз связана с изменением степени окисления церия из +4 в +3 в процессе нагрева [123–126] и образованием в качестве конечного продукта CePO4.
Два других изоструктурных двойных ортофосфата церия(IV) ((NH4)2Ce(PO4)2 ⋅ H2O и K2Ce(PO4)2 ⋅ H2O) были получены Ху и др. [127, 128] гидротермальным методом из смеси гидратированного CeO2 и частично нейтрализованной ортофосфорной кислоты. Была проведена оценка протонной проводимости полученных соединений и показано, что (NH4)2Ce(PO4)2 ⋅ H2O сохраняет протонную проводимость даже после удаления воды из структуры, что, вероятно, обусловлено участием катионов аммония в переносе заряда.
Сальвадо и др. [129] указали на то, что Ху и др. неправильно описали структуру (NH4)2Ce(PO4)2 ⋅ ⋅ H2O, и на самом деле это соединение имеет совершенно другие параметры элементарной ячейки, близкие к параметрам изоструктурного (NH4)2Th(PO4)2 ⋅ H2O [116]. Структура (NH4)2Ce(PO4)2 ⋅ H2O, как и других двойных ортофосфатов церия(IV), фактически представляет собой трехмерный каркас и содержит туннели разных размеров. Катионы аммония и молекулы воды при этом находятся в туннелях большего размера (рис. 5). Шекунова и др. [115] синтезировали аналогичное соединение гидротермальной обработкой аморфного геля ортофосфата церия(IV) в среде 2.5‒3 М водного раствора аммиака.
Рис. 5.
Структура (NH4)2Ce(PO4)2 ⋅ H2O вдоль оси a. Зеленым цветом обозначены полиэдры CeO8, синим – тетраэдры PO4, голубым и красным цветом показаны молекулы воды и аммиака соответственно [129].
Брэгиру и др. [130] систематизировали имеющиеся сведения о семействе двойных ортофосфатов общего состава MIIḾIV(PO4)2 (MII = Cd, Ca, Sr, Pb, Ba; ḾIV = Ge, Ti, Mo, Sn, Hf, Zr, Pu, Np, U, Th). Было показано, что в зависимости от ионных радиусов MII и ḾIV соединения MIIḾIV(PO4)2 (за некоторыми исключениями) кристаллизуются в структурных типах явапаита и чералита. Для соединений такого состава, где в качестве ḾIV выступал бы церий, имеются неподтвержденные литературные данные о получении MIICeIV(PO4)2 (MII = Mg, Ca, Sr, Ba, Cd) [111], а также сведения о возможном существовании твердых растворов Ce2 – 2xBaxḾIV(PO4)2 (ḾIV = Zr, Hf; x < 0.2) со структурой типа чералита [131].
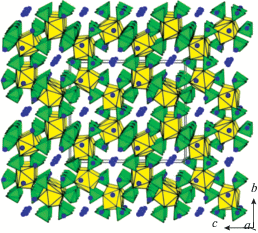
Высокотемпературный гидротермальный синтез был использован авторами [132] для получения первого ортофосфата церия(IV) Na10Ce2P6O24 (Na10Ce2(PO4)6) с мольным отношением P/Ce = 3 и двойного ортофосфата урана(IV) аналогичного состава. Соединение состава Na5Ce(PO4)3 с аналогичной дифрактограммой было описано ранее [133], однако для него не была определена структура. Лай и др. [132] показали, что структуры Na10Ce2P6O24 и Na10U2P6O24 имеют каркасное строение с пересекающимися туннелями вдоль осей a и b, в которых находятся катионы натрия. Трехмерный каркас образован полиэдрами MO8 (М = Ce, U) и тетраэдрами PO4, сочлененными вершинами и ребрами (рис. 6).
Рис. 6.
Структура Na10Ce2P6O24. Желтым цветом обозначены полиэдры CeO8, зеленым – тетраэдры РО4, синим – атомы натрия [132].
Несмотря на то, что гидротермальный метод наиболее часто используется для получения двойных ортофосфатов церия(IV), известны и другие подходы к их синтезу.
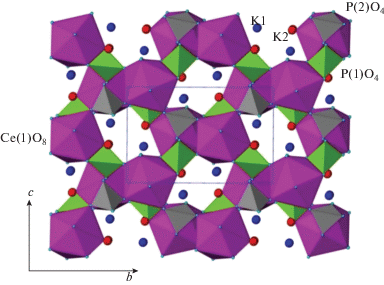
Бевара и др. [134] твердофазным синтезом исходя из KH2PO4 и CeO2 получили соединение состава K2Ce(PO4)2, структура которого представляет собой трехмерный каркас, образованный полиэдрами CeO8 и тетраэдрами РО4, с катионами калия внутри туннелей (рис. 7). Авторы [135] продемонстрировали эффективность ионного обмена катионов калия на катионы стронция в широком диапазоне рН, при этом максимальное извлечение Sr2+ наблюдалось при рН 14.
Рис. 7.
Структура K2Ce(PO4)2. Розовым цветом обозначены полиэдры CeO8, зеленым и серым – тетраэдры PO4. Изолированными сферами синего и красного цвета обозначены катионы калия, маленькими голубыми сферами – атомы кислорода [134].
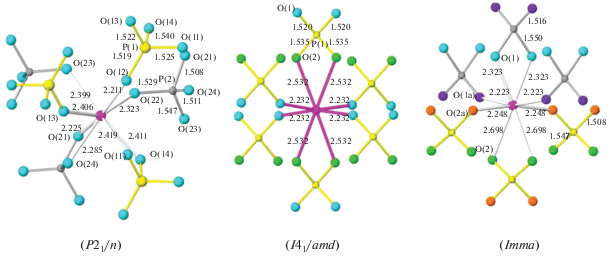
Анализ термического разложения K2Ce(PO4)2 показал, что конечным продуктом термолиза этой фазы при температурах около 900°С является смесь CePO4 и K3Ce(PO4)2 [134]. Детальное исследование процессов, происходящих при более низких температурах, позволило авторам [136] обнаружить новые структурные модификации исходного соединения. Оказалось, что при температурах ~500°С происходит фазовый переход с изменением локального окружения CeO8 и PO4, вследствие чего исходная моноклинная сингония переходит в ромбическую, а затем в тетрагональную (рис. 8). Объем элементарной ячейки высокотемпературной фазы на ~14.4% больше, чем объем элементарной ячейки исходной фазы K2Ce(PO4)2, кристаллизующейся в моноклинной сингонии. Примечательно, что высокотемпературная модификация с тетрагональной сингонией при охлаждении может обратимо переходить в низкотемпературную модификацию с моноклинной сингонией.
Рис. 8.
Локальная структура K2Ce(PO4)2 в моноклинной (P21/n), тетрагональной (I41/amd) и ромбической (Imma) сингониях (числами обозначены длины связей) [136].
Бевара и др. [137] выполнили также аналогичное исследование термического разложения K2Th(PO4)2 с учетом изоструктурности этой фазы с K2Ce(PO4)2. В целом, происходящие при нагревании этой фазы процессы подобны процессам термолиза K2Ce(PO4)2, хотя имеются и определенные различия (например, разница коэффициентов термического расширения), обусловленные разной длиной связей M–O (M = Th, Ce).
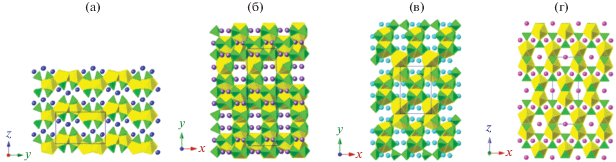
Ю и др. [138] твердофазным синтезом получили четыре фазы общего состава A2Th(PO4)2 (A = = Li, Na, Rb, Cs). Показано, что структуру продукта определяет катион. Так, Li2Th(PO4)2, Na2Th(PO4)2 и K2Th(PO4)2 имели различные параметры элементарной ячейки, в отличие от изоструктурных Rb2Th(PO4)2 и Cs2Th(PO4)2. Более того, фаза Li2Th(PO4)2 имела слоистое строение, в отличие от остальных четырех соединений, которыe характеризовались наличием трехмерного каркаса (рис. 9).
Рис. 9.
Кристаллические структуры Li2Th(PO4)2 (а), Na2Th(PO4)2 (б), K2Th(PO4)2 (в), Rb2Th(PO4)2 и Cs2Th(PO4)2 (г). Полиэдры, содержащие Th и тетраэдры PO4, обозначены желтым и зеленым цветом соответственно. Атомы Li обозначены голубым цветом, атомы Na ‒ фиолетовым, атомы K ‒ светло-голубым, атомы Rb и Cs ‒ розовым [138].
Огородник и др. [139] сплавлением KPO3, K4P2O7, ZrF4 и CeF4 получили тройной ортофосфат церия(IV) состава K4CeZr(PO4)4, структура которого представляет трехмерный каркас, образованный полиэдрами ZrO6, CeO8 и тетраэдрами PO4. Катионы калия занимают туннели, расположенные вдоль осей а и b. Отметим, что сравнение литературных данных указывает на отличие структур ортофосфатов циркония (см., например, работы [140–142]) от структур ортофосфатов церия и актинидов, что, по-видимому, объясняется различными координационными полиэдрами, характерными для этих металлов (ZrO6 и Сe(Ac)O8).
КРИСТАЛЛИЧЕСКИЕ ОРТОФОСФАТЫ ЦЕРИЯ(IV): СМЕШАННЫЕ СОЛИ
Количество известных на сегодняшний день смешанных ортофосфатов церия(IV) и изоструктурных им соединений сравнительно невелико. Кониг и Экстайн [143] получили кристаллический гидрофосфат-сульфат церия(IV) состава Ce2O(HPO4)3 –x(SO4)x ⋅ 4H2O (0 < x < 1). Была продемонстрирована способность полученного соединения к ионному обмену, однако не были представлены структурные и иные данные, подтверждающие правильность определения химического состава продукта.
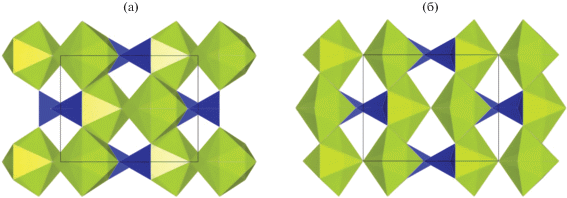
Некоторые авторы сообщали о получении гидроксоортофосфата церия(IV) состава Ce(OH)PO4. Так, Херман и Клирфилд [94] заявили о синтезе семейства соединений состава Ce(OH)x(PO4)x(HPO4)2 – 2x, включающем фазу Ce(OH)PO4 (x = 1), однако определенный ими брутто-состав конечного продукта соответствовал формуле Ce(OH)0.7(PO4)1.1. Хаяши и др. [46] гидротермальной обработкой аморфного Ce(HPO4)2 ⋅ nH2O синтезировали соединение, идентичное, по данным рентгенофазового анализа, продукту, полученному Херманом и Клирфилдом [94]. Лебедев и Руденко [144] предположили возможность образования Ce(OH)PO4 в качестве промежуточного соединения при синтезе CeO2. На вероятность формирования Ce(OH)PO4 в результате нагревания CePO4 ⋅ nH2O на воздухе было указано и в классической работе И.В. Тананаева и др. [57]. Тем не менее до недавнего времени не было представлено убедительных данных о составе и структуре соединения с формулой Ce(OH)PO4. Только в 2020 г. Козлова и др. [145] предложили способ получения и определили структуру данного соединения. Авторы указали, что синтез Ce(OH)PO4 в водной среде представляет собой сложную задачу ввиду высокой скорости гидролиза церия(IV) даже в сильнокислых средах [146], вследствие этого ранее был известен лишь один пример основной соли церия(IV) ‒ Сe2(OH)2(H2O)4(SO4)3 [147]. Синтез Ce(OH)PO4 осуществляли гидротермальной обработкой аморфного церийфосфатного геля в среде 3 М азотной кислоты [145]. Было показано, что полученный гидроксоортофосфат церия(IV) изоструктурен соединениям тория и урана Th(OH)PO4 и U(OH)PO4 [148]. Необходимо отметить, что вопрос о присутствии в составе U(OH)PO4 кристаллизационной воды [148, 149] вызывал дискуссию вплоть до 2019 г., когда достоверно было доказано ее отсутствие [150].
Авторами [145] было установлено, что термолиз Ce(OH)PO4 сопровождается формированием новой фазы с близкой к исходному соединению структурой. Анализ структурных данных позволил определить, что продуктом термического разложения Ce(OH)PO4 при ~300°С является оксоортофосфат церия(IV) Ce2O(PO4)2 (рис. 10). Данное соединение изоструктурно Th2O(PO4)2 [148], U2O(PO4)2, Np2O(PO4)2 [151] и β-Zr2O(PO4)2 [152, 153]. Отметим, что высокотемпературную модификацию Hf2O(PO4)2 также считали изоструктурной β-Zr2O(PO4)2 [154], однако эта гипотеза была впоследствии опровергнута [155].
Рис. 10.
Структуры Ce(OH)PO4 (а) и Ce2O(PO4)2 (б) вдоль оси с. Полиэдры CeOn (n = 8 и 7 для Ce(OH)PO4 и Ce2O(PO4)2 соответственно) обозначены желто-зеленым цветом, атомы фосфора находятся внутри тетраэдров PO4, обозначенных синим цветом [145].
Полученные Козловой и др. [145] результаты позволяют предположить возможность получения Pu2O(PO4)2 аналогичным способом.
В обзоре Брандела и др. [151] отмечается, что гидроксоортофосфат урана(IV) способен к анионному обмену при контакте с растворами соляной или бромистоводородной кислоты. Бенардом и др. [156] были получены два изоструктурных друг другу соединения состава UXPO4 · 2H2O (X = Cl, Br) растворением металлического урана в концентрированных растворах HCl и HBr, соответственно, с последующим взаимодействием растворов с 15 М H3PO4 (U/PO4 = 1). Отметим, что для церия аналогичные соединения к настоящему времени не описаны.
Вызывает определенные сомнения и возможность получения Ce4(PO4)4P2O7, обладающего, по предположениям некоторых авторов [96, 157], структурой, близкой к Th4(PO4)4P2O7 [99, 158].
В работе [159] гидротермальной обработкой смешанного раствора Ce(SO4)2, H3PO4, H2NCH2CH2NH2 и NH4F был синтезирован ортофосфат-фторид церия(IV) (NH4)[CeF2(PO4)] и показано, что структура этого соединения образована полиэдрами CeO4F4 и тетраэдрами PO4, составляющими трехмерный каркас с катионами аммония внутри каналов (рис. 11а).
Помимо этого Ю и др. [160] в гидротермальных условиях получили слоистый гидроортофосфат-фторид церия(IV) [(CH2)2(NH3)2]0.5[CeF3(HPO4)], состоящий из слоев, образованных полиэдрами CeO3F5 и PO4, с органическим катионом [NH3(CH2)2NH3]2+, находящимся между ними (рис. 11б).
Изоструктурные ортофосфатам-фторидам церия(IV) соединения актинидов пока не описаны. Брандел и др. [161] упоминали о возможном получении фазы состава ThFPO4 · H2O гидротермальной обработкой смеси Th4+–H3PO4–HF, однако ими не были приведены подтверждающие структурные данные. Фелдер и др. [162] в 2018 г. предложили способ синтеза UFPO4 в гидротермальных условиях исходя из смеси ацетата уранила, ортофосфорной кислоты и фторида железа(III) и определили его структуру. Было показано, что продукт синтеза изоструктурен полученному ранее NpFPO4 [163]. Аналогичное соединение церия(IV) CeFPO4 в литературе не описано, однако Леблан и др. сообщают об изоструктурности NpFPO4 и CeFAsO4 [164]. Следует отметить, что в качестве структурных аналогов ортофосфатов зачастую рассматривают именно арсенаты [30, 119, 138, 165–167]. Действительно, параметры элементарной ячейки CeFAsO4 [168] оказались близки к параметрам ячейки NpFPO4. Помимо CeFAsO4 Роуз и Веллер [168] синтезировали в гидротермальных условиях три других арсената-фторида церия(IV) состава [(NH4)5(H2O)2][Ce4(AsO4)6(H2O)F3], Ce[AsO4]F[H2O] и (NH4)[CeF2(AsO4)], используя в качестве реагентов CeF4, NH4H2AsO4 или H3AsO4. Было показано, что фаза [(NH4)5(H2O)2] [Ce4(AsO4)6(H2O)F3] характеризуется каркасом, образованным двумя типами полиэдров (CeO7F и CeO7OH2) и тетраэдрами AsO4, с катионами ${\text{NH}}_{4}^{ + }$ и молекулами воды внутри больших туннелей. Структуры Ce[AsO4]F и Ce[AsO4]F[H2O] образованы полиэдрами CeO6F2 и CeO5F2(OH2) соответственно, соединенными друг с другом связями Ce‒O‒Ce и Ce‒F‒Ce (рис. 12).
Рис. 12.
Структуры: а – [(NH4)5(H2O)2][Ce4(AsO4)6(H2O)F3] (светло-серые полиэдры ‒ CeO7F, темно-серые полиэдры ‒ CeO7OH2, серые тетраэдры ‒ AsO4, белые сферы – молекулы H2O, черные сферы – катионы NH4+), б – Ce[AsO4]F (серые полиэдры ‒ CeO6F2, темно-серые тетраэдры ‒ AsO4, белые сферы – O2–, серые сферы – F–), в – Ce[AsO4]F[H2O] (серые полиэдры ‒ CeO5F2(ОН2), темно-серые тетраэдры ‒ AsO4, белые сферы – O2–, серые сферы – F–) вдоль оси b [168].
Соединение (NH4)[CeF2(AsO4)] изоструктурно (NH4)[CeF2(PO4)], полученному в [159] и описанному выше. На основании вышеизложенного можно предположить возможность получения изоструктурных соединений церия(IV) [(NH4)5(H2O)2][Ce4(PO4)6(H2O)F3], Ce[PO4]F[H2O] и Ce[PO4]F.
ЗАКЛЮЧЕНИЕ
Настоящий обзор представляет собой первую попытку обобщить имеющиеся сведения об ортофосфатах церия(IV), включая аморфные и кристаллические гидроортофосфаты, двойные и смешанные соли церия(IV). Гидроортофосфаты церия(IV) зачастую формируются в аморфном виде, в том числе в виде гелей, которые характеризуются уникальной одномерной микроструктурой. Состав аморфных гидроортофосфатов церия(IV) и причины формирования особой структуры этих соединений до сих пор надежно не установлены. Аморфные гидроортофосфаты церия(IV), в том числе в составе полимерных композитов, рассматривают в качестве перспективных ионообменных материалов и сорбентов. Кристаллические гидроортофосфаты церия(IV) в настоящее время представлены всего двумя соединениями с достоверно охарактеризованной структурой – Ce2(PO4)2HPO4 ⋅ ⋅ H2O и Ce(PO4)1.5(H2O)(H3O)0.5(H2O)0.5. Семейство кристаллических двойных и смешанных ортофосфатов церия(IV), сведения о которых подтверждены рентгеноструктурными данными, насчитывает на сегодняшний день 11 соединений. Область практического использования кристаллических ортофосфатов церия(IV) включает иммобилизацию радиоактивных отходов, возможную благодаря ионообменным свойствам этих соединений, а также близости ионных радиусов церия(IV) и актинидов.
Анализ литературных данных, посвященных в первую очередь кристаллическим ортофосфатам актинидов и металлов 4 группы, позволил провести структурные аналогии с ортофосфатами церия(IV). В частности, описаны изоструктурные соединения Ce2(PO4)2HPO4 ⋅ H2O и Th2(PO4)2HPO4 · ⋅ H2O; (NH4)2Ce(PO4)2 ⋅ H2O и (NH4)2Th(PO4)2 ⋅ ⋅ H2O; NH4Ce2(PO4)3 и NH4Th2(PO4)3; K2Ce(PO4)2 и K2Th(PO4)2; Ce(OH)PO4 и An(OH)PO4 (An = Th, U); Ce2O(PO4)2 и M2O(PO4)2 (M = Th, U, Np, Zr) и др. Поскольку класс ортофосфатов актинидов существенно шире класса ортофосфатов церия(IV), структурное подобие этих фаз позволяет предположить существование ряда новых изоструктурных соединений четырехвалентного церия. В свою очередь, представленные сведения об ортофосфатах церия(IV) могут быть полезны для предсказания возможности синтеза новых ортофосфатов актинидов, в том числе Pu(IV) и Np(IV).
Список литературы
Behrsing T., Deacon G.B., Junk P.C. // The chemistry of rare earth metals, compounds, and corrosion inhibitors. Amsterdam: Elsevier Ltd, 2015. https://doi.org/10.1533/9780857093585.1
Cotton S. // Lanthanide and Actinide Chemistry. Chichester: John Wiley & Sons Ltd, 2006. https://doi.org/10.1002/0470010088
Voncken J.H.L. // The Rare Earth Elements. Cham: Springer International Publishing, 2016. https://doi.org/10.1007/978-3-319-26809-5
Migaszewski Z.M., Gałuszka A. // Crit. Rev. Environ. Sci. Technol. 2015. V. 45. № 5. P. 429. https://doi.org/10.1080/10643389.2013.866622
Соболев Б.П. // Журн. неорган. химии. 2020. Т. 65. № 3. С. 373. [Soblev B.P. // Russ. J. Inorg. Chem. 2020. V. 65. № 3. P. 395. https://doi.org/10.1134/S0036023620030158]https://doi.org/10.31857/S0044457X20030150
Ochiai A., Utsunomiya S. // Minerals. 2017. V. 7. № 84. P. 1. https://doi.org/10.3390/min7050084
Krishnamurthy N., Gupta C.K. // Extractive Metallurgy of Rare Earths. Boca Raton: Taylor & Francis Group, 2016.
Balaram V. // Geosci. Front. 2019. V. 10. № 4. P. 1285. https://doi.org/10.1016/j.gsf.2018.12.005
Yan C.H., Yan Z.G., Du Y.P. et al. // Handb. Phys. Chem. Rare Earths. Amsterdam: Elsevier Ltd, 2011. P. 275. https://doi.org/10.1016/B978-0-444-53590-0.00004-2
Li Q., Yam V.W.W. // Angew. Chem. Int. Ed. 2007. V. 46. № 19. P. 3486. https://doi.org/10.1002/anie.200604973
Lv C., Di W., Liu Z. et al. // Analyst. 2014. V. 139. № 18. P. 4547. https://doi.org/10.1039/C4AN00952E
Di W., Wang X., Ren X. // Nanotechnology. 2010. V. 21. № 7. P. 075709. https://doi.org/10.1088/0957-4484/21/7/075709
Norby T., Christiansen N. // Solid State Ionics. 1995. V. 77. P. 240. https://doi.org/10.1016/0167-2738(94)00274-V
Onoda H., Nariai H., Moriwaki A. et al. // J. Mater. Chem. 2002. V. 12. № 6. P. 1754. https://doi.org/10.1039/b110121h
Neumeier S., Arinicheva Y., Ji Y. et al. // Radiochim. Acta. 2017. V. 105. № 11. P. 961. https://doi.org/10.1515/ract-2017-2819
Lucas S., Champion E., Bregiroux D. et al. // J. Solid State Chem. 2004. V. 177. № 4–5. P. 1302. https://doi.org/10.1016/j.jssc.2003.11.003
Lucas S., Champion E., Bernache-Assollant D. et al. // J. Solid State Chem. 2004. V. 177. № 4–5. P. 1312. https://doi.org/10.1016/j.jssc.2003.11.004
Onoda H., Nariai H., Maki H. et al. // Mater. Chem. Phys. 2003. V. 78. № 2. P. 400. https://doi.org/10.1016/S0254-0584(02)00208-0
Patra C.R., Alexandra G., Patra S. et al. // New J. Chem. 2005. V. 29. № 5. P. 733. https://doi.org/10.1039/b415693e
Yang R., Qin J., Li M. et al. // CrystEngComm. 2011. V. 13. № 24. P. 7284. https://doi.org/10.1039/c1ce05368j
Скогарева Л.С., Шекунова Т.О., Баранчиков А.Е. и др. // Журн. неорган. химии. 2016. Т. 61. № 10. С. 1276. [Skogareva L.S., Shekunova T.O., Baranchikov A.E. et al. // Russ. J. Inorg. Chem. 2016. V. 61. № 10. P. 1219. https://doi.org/10.1134/S0036023616100181]https://doi.org/10.7868/s0044457x16100184
Clavier N., Podor R., Dacheux N. // J. Eur. Ceram. Soc. 2011. V. 31. № 6. P. 941. https://doi.org/10.1016/j.jeurceramsoc.2010.12.019
Mooney R.C.L. // Acta Crystallogr. 1950. V. 3. № 5. P. 337. https://doi.org/10.1107/s0365110x50000963
Mesbah A., Clavier N., Elkaim E. et al. // Cryst. Growth Des. 2014. V. 14. № 10. P. 5090. https://doi.org/10.1021/cg500707b
Mesbah A., Clavier N., Elkaim E. et al. // J. Solid State Chem. 2017. V. 249. P. 221. https://doi.org/10.1016/j.jssc.2017.03.004
Schlenz H., Heuser J., Neumann A. et al. // Z. Kristallogr. 2013. V. 228. № 3. P. 113. https://doi.org/10.1524/zkri.2013.1597
Boatner L.A. // Rev. Mineral. Geochemistry. 2002. V. 48. № 1. P. 87. https://doi.org/10.2138/rmg.2002.48.4
Shelyug A., Mesbah A., Szenknect S. et al. // Front. Chem. 2018. V. 6. P. 1. https://doi.org/10.3389/fchem.2018.00604
Rafiuddin M.R., Grosvenor A.P. // Inorg. Chem. 2016. V. 55. № 19. P. 9685. https://doi.org/10.1021/acs.inorgchem.6b01471
Achary S.N., Bevara S., Tyagi A.K. // Coord. Chem. Rev. 2017. V. 340. № March. P. 266. https://doi.org/10.1016/j.ccr.2017.03.006
Sroor F.M.A., Edelmannand F.T. // Rare Earth Elem. Fundam. Appl. / Ed. Atwood D.A., Chichester: John Wiley & Sons Ltd, 2012. P. 313.
Plakhova T.V., Romanchuk A.Y., Yakunin S.N. et al. // J. Phys. Chem. C. 2016. V. 120. № 39. P. 22615. https://doi.org/10.1021/acs.jpcc.6b05650
Boyd C.E. // Water Quality, 3rd ed., Springer International Publishing, Cham, 2020. https://doi.org/10.1007/978-3-030-23335-8
Wagh A.S. // Twenty-First Century Materials with Diverse Applications. Amsterdam: Elsevier Ltd, 2016.
Кочетков С.П., Смирнов Н.Н., Ильин А.П. // Концентрирование и очистка экстракционной фосфорной кислоты. Иваново: ГОУВПО Иван. гос. хим.-технол. ун-т, 2007.
Korte C., Conti F., Wackerl J. et al. High Temp. Polym. Electrolyte Membr. Fuel Cells Approaches, Status, Perspect / Eds. Li Q. et al. Switzerland, Springer International Publishing, 2016. P. 169.
Лебедев И.А., Куляко Ю.М. // Журн. неорган. химии. 1978. V. 23. № 12. P. 3215.
König K.-H., Meyn E. // J. Inorg. Nucl. Chem. 1967. V. 29. № 4. P. 1153. https://doi.org/10.1016/0022-1902(67)80101-5
Byrne R.H., Lee J.H., Bingler L.S. // Geochim. Cosmochim. Acta. 1991. V. 55. P. 2729. https://doi.org/10.1016/0016-7037(91)90439-C
Чиркст Д.Э., Черемисина О.В. // Записки Горного института 2006. V. 169. P. 219. https://pmi.spmi.ru/index.php/pmi/article/view/7890/5815
Marsac R., Réal F., Banik N.L. et al. // Dalton Trans. 2017. V. 46. № 39. P. 13553. https://doi.org/10.1039/c7dt02251d
Elyahyaoui A., Brillard L., Boulhassa S. et al. // Radiochim. Acta. 1990. V. 49. P. 39. https://doi.org/10.1524/ract.1990.49.1.39
Hartley W.N. // J. Chem. Soc. 1882. V. 41. P. 202. https://doi.org/10.1039/CT8824100202
Alberti G., Constantino U., Di Gregorio F. et al. // J. Inorg. Nucl. Chem. 1968. V. 30. № 1. P. 295. https://doi.org/10.1016/0022-1902(68)80093-4
Del Rey-Bueno F., Villafranca-Sanchez E., Mata-Arjona A. et al. // Mater. Chem. Phys. 1989. V. 21. № 1. P. 49. https://doi.org/10.1016/0254-0584(89)90101-6
Hayashi H., Ebina T., Onodera Y. et al. // Bull. Chem. Soc. Jpn. 1997. V. 70. P. 1701. https://doi.org/10.1246/bcsj.70.1701
Alberti G., Costantino U. // J. Chromatogr. 1970. V. 50. P. 482. https://doi.org/10.1016/S0021-9673(00)97976-7
Perez-Jimenez C., Villafranca-Sanchez E., Gonzalez-Pradas E. et al. // Mater. Chem. Phys. 1987. V. 15. P. 411. https://doi.org/10.1016/0254-0584(87)90061-7
Barboux P., Morineau R., Livage J. // Solid State Ionics. 1988. V. 27. № 4. P. 221. https://doi.org/10.1016/0167-2738(88)90213-5
Casciola M., Costantino U., D’Amico S. // Solid State Ionics. 1988. V. 28–30. P. 617. https://doi.org/10.1016/S0167-2738(88)80112-7
Ishii K., Kimura Y., Yamazaki T. et al. // RSC Adv. 2017. V. 7. № 57. P. 35711. https://doi.org/10.1039/c7ra06850f
Mondal C., Ganguly M., Pal J. et al. // Chem. Commun. 2013. V. 49. № 82. P. 9428. https://doi.org/10.1039/c3cc45555f
Yorov K.E., Shekunova T.O., Baranchikov A.E. et al. // J. Sol-Gel Sci. Technol. 2018. V. 85. № 3. P. 574. https://doi.org/10.1007/s10971-018-4584-3
Livage J. // Chem. Mater. 1991. V. 3. № 4. P. 578. https://doi.org/10.1021/cm00016a006
Kozlova T.O., Baranchikov A.E., Kozlov D.A. et al. // ACS Omega. 2020. V. 5. № 28. P. 17592. https://doi.org/10.1021/acsomega.0c02061
Baes C.F., Mesmer R.S. // The Hydrolysis of Cations. New York, London, Sydney, Toronto: John Wiley & Sons, 1976.
Тананаев И.В., Розанов И.А., Авдуевская К.А. и др. // Фосфаты четырехвалентных элементов. М.: Наука, 1972.
Inoue Y. // Bull. Chem. Soc. Jpn. 1963. V. 36. № 10. P. 1316. https://doi.org/10.1246/bcsj.36.1316
Parmar M.U., Mehta S.M., Prasad M. // J. Indian Chem. Soc. 1935. P. 107. https://doi.org/10.1007/BF03035821
Amarantov S.V., Shekunova T.O., Baranchikov A.E. et al. // J. Surf. Invest. X-Ray, Synchrotron Neutron Tech. 2020. V. 14. P. S201. https://doi.org/10.1134/S1027451020070034
Rocco G.G., Weiner J.R., Cali J.P. // Phys. Sci. Res. Pap. 1964. V. 73. P. 1.
Larsen E., Cilley W. // J. Inorg. Nucl. Chem. 1968. V. 30. № 1. P. 287. https://doi.org/10.1016/0022-1902(68)80092-2
Alberti G., Costantino U. // J. Chromatogr. 1974. V. 102. P. 5. https://doi.org/10.1016/S0021-9673(01)85423-6
Hayashi H., Iwasaki T., Nagase T. et al. // Solvent Extr. Ion Exch. 1995. V. 13. № 6. P. 1145. https://doi.org/10.1080/07366299508918322
Alberti G., Casciola M., Costantino U. et al. // J. Chromatogr. 1976. V. 128. P. 289. https://doi.org/10.1016/S0021-9673(00)99260-4
Clearfield A. // Inorganic ion exchange materials. Boca Raton: Taylor & Francis Group, 2018. https://doi.org/10.1201/9781351073561
Alberti G., Massucci M.A., Torracca E. // J. Chromatogr. 1967. V. 30. P. 579. https://doi.org/10.1016/S0021-9673(00)84193-X
De A.K., Chowdhury K. // J. Chromatogr. 1974. V. 101. P. 63. https://doi.org/10.1016/S0021-9673(01)94731-4
Романчук А.Ю., Шекунова Т.О., Петров В.Г. и др. // Радиохимия. 2018. Т. 60. № 6. С. 525. [Romanchuk A.Y., Shekunova T.O., Petrov V.G. et al. // Radiochemistry. 2018. V. 60. № 6. P. 613. https://doi.org/10.1134/S1066362218060085]https://doi.org/10.1134/s0134347518060086
Varshney K.G., Rafiquee M.Z.A., Somya A. // J. Therm. Anal. Calorim. 2007. V. 90. № 3. P. 663. https://doi.org/10.1007/s10973-007-8519-4
Iqbal N., Mobin M., Rafiquee M.Z.A. // Chem. Eng. J. 2011. V. 169. № 1–3. P. 43. https://doi.org/10.1016/j.cej.2011.02.048
Iqbal N., Mobin M., Rafiquee M.Z.A. et al. // Desalin. Water Treat. 2013. V. 51. № 34–36. P. 6688. https://doi.org/10.1080/19443994.2013.765809
Varshney K.G., Rafiquee M.Z.A., Somya A. // Colloids Surf., A: Physicochem. Eng. Asp. 2007. V. 301. № 1–3. P. 69. https://doi.org/10.1016/j.colsurfa.2006.12.025
Somya A., Rafiquee M.Z.A., Varshney K.G. // Colloids Surf., A: Physicochem. Eng. Asp. 2009. V. 336. № 1–3. P. 142. https://doi.org/10.1016/j.colsurfa.2008.11.036
Varshney K.G., Rafiquee M.Z.A., Somya A. // Colloids Surf., A: Physicochem. Eng. Asp. 2008. V. 317. № 1–3. P. 400. https://doi.org/10.1016/j.colsurfa.2007.11.012
Somya A., Rafiquee M.Z.A., Varshney K.G. et al. // Desalin. Water Treat. 2015. V. 56. № 3. P. 638. https://doi.org/10.1080/19443994.2014.940388
Iqbal N., Mobin M., Rafiquee M.Z.A. et al. // Desalin. Water Treat. 2016. V. 57. № 42. P. 19917. https://doi.org/10.1080/19443994.2015.1109556
Iqbal N., Mobin M., Rafiquee M.Z.A. et al. // Chem. Eng. Res. Des. 2012. V. 90. № 12. P. 2364. https://doi.org/10.1016/j.cherd.2012.06.006
Iqbal N., Rafiquee M.Z.A. // Colloids Surf., A: Physicochem. Eng. Asp. 2010. V. 364. № 1–3. P. 67. https://doi.org/10.1016/j.colsurfa.2010.04.039
El-Azony K.M., Ismail Aydia M., El-Mohty A.A. // J. Radioanal. Nucl. Chem. 2011. V. 289. № 2. P. 381. https://doi.org/10.1007/s10967-011-1079-x
Varshney K.G., Tayal N., Gupta U. // Colloids Surf., A: Physicochem. Eng. Asp. 1998. V. 145. № 1–3. P. 71. https://doi.org/10.1016/S0927-7757(98)00657-8
Varshney K.G., Gupta P., Tayal N. // Indian J. Chem., Sect. A: Inorg., Phys. Theor. Anal. Chem. 2003. V. 42. № 1. P. 89. http://nopr.niscair.res.in/handle/123456789/20501
Varshney K.G., Agrawal A., Mojumdar S.C. // J. Therm. Anal. Calorim. 2005. V. 81. P. 183. https://doi.org/10.1007/s10973-005-0765-8
Metwally S.S., El-Gammal B., Aly H.F. et al. // Sep. Sci. Technol. 2011. V. 46. № 11. P. 1808. https://doi.org/10.1080/01496395.2011.572328
El-Gammal B., Metwally S.S., Aly H.F. et al. // Desalin. Water Treat. 2012. V. 46. № 1–3. P. 124. https://doi.org/10.1080/19443994.2012.677412
Shakshooki S.K., El-Akari F.A., El-Fituri S.M. et al. // Adv. Mater. Res. 2014. V. 856. P. 3. https://doi.org/10.4028/www.scientific.net/AMR.856.3
Herman R.G., Clearfield A. // J. Inorg. Nucl. Chem. 1976. V. 38. P. 853. https://doi.org/10.1016/0022-1902(76)80370-3
Nazaraly M., Chanéac C., Ribot F. et al. // J. Phys. Chem. Solids. 2007. V. 68. P. 795. https://doi.org/10.1016/j.jpcs.2007.03.010
Nazaraly M., Quarton M., Wallez G. et al. // Solid State Sci. 2007. V. 9. P. 672. https://doi.org/10.1016/j.solidstatesciences.2007.04.021
Tsuhako M., Danjo M., Baba Y. et al. // Bull. Chem. Soc. Jpn. 1997. V. 70. № 1. P. 143. https://doi.org/10.1246/bcsj.70.143
Nazaraly M., Wallez G., Chanéac C. et al. // Angew. Chem. Int. Ed. 2005. V. 44. P. 5691. https://doi.org/10.1002/anie.200501871
Hayashi H., Torii K., Nakata S.I. // J. Mater. Chem. 1997. V. 7. № 3. P. 557. https://doi.org/10.1039/a606397g
Shekunova T.O., Baranchikov A.E., Ivanova O.S. et al. // J. Non. Cryst. Solids. 2016. V. 447. P. 183. https://doi.org/10.1016/j.jnoncrysol.2016.06.012
Herman R.G., Clearfield A. // J. Inorg. Nucl. Chem. 1975. V. 37. P. 1697. https://doi.org/10.1016/0022-1902(75)80301-0
Романчук А.Ю., Шекунова Т.О., Ларина А.И. и др. // Радиохимия. 2019. Т. 61. № 6. С. 512. [Romanchuk A.Y., Shekunova T.O., Larina A.I. et al. // Radiochemistry. 2019. V. 61. № 6. P. 719. https://doi.org/10.1134/S1066362219060134]https://doi.org/10.1134/s00338311190600121
Brandel V., Clavier N., Dacheux N. // J. Solid State Chem. 2005. V. 178. № 4. P. 1054. https://doi.org/10.1016/j.jssc.2005.01.005
Sato T., Sato C., Yin S. // Phosphorus Res. Bull. 2008. V. 22. P. 17. https://doi.org/10.3363/prb.22.17
Sato T., Yin S. // Phosphorus Res. Bull. 2010. V. 24. P. 43. https://doi.org/10.3363/prb.24.43
Dacheux N., Clavier N., Wallez G. et al. // Mater. Res. Bull. 2005. V. 40. № 12. P. 2225. https://doi.org/10.1016/j.materresbull.2005.06.011
Salvadó M.A., Pertierra P., Bortun A.I. et al. // Inorg. Chem. 2005. V. 44. № 10. P. 3512. https://doi.org/10.1021/ic048216f
Dacheux N., Grandjean S., Rousselle J. et al. // Inorg. Chem. 2007. V. 46. № 24. P. 10390. https://doi.org/10.1021/ic701297v
Troup J.M., Clearfield A. // Inorg. Chem. 1977. V. 16. № 12. P. 3311. https://doi.org/10.1021/ic50178a065
Pet’kov V.I. // Russ. Chem. Rev. 2012. V. 81. № 7. P. 606. https://doi.org/10.1070/rc2012v081n07abeh004243
Poojary D.M., Shpeizer B., Clearfield A. // J. Chem. Soc., Dalton Trans. 1995. № 1. P. 111. https://doi.org/10.1039/DT9950000111
Salvadó M.A., Pertierra P., Castro G.R. et al. // Inorg. Chem. 2008. V. 47. № 4. P. 1246. https://doi.org/10.1021/ic702497x
Borhan A., Apetrăchioaei B., Popa K. // Rev. Roum. Chim. 2010. V. 55. № 7. P. 389. http://revroum.lew.ro/wp-content/uploads/2010/RRCh_7_2010/ Art%2006.pdf
Bamberger C.E., Haire R.G., Begun G.M. et al. // J. Less-Common Met. 1984. V. 102. P. 179. https://doi.org/10.1016/0022-5088(84)90314-X
Orlova A.I., Kitaev D.B. // Radiochemistry. 2005. V. 47. № 1. P. 14. [Орлова А.И., Китаев Д.Б. // Радиохимия. 2005. Т. 47. № 1. С. 15.]https://doi.org/10.1007/s11137-005-0041-6
Orlova A.I., Kitaev D.B., Volkov Yu.F. et al. // Radiochemistry. 2001. V. 43. № 3. P. 225. [Орлова А.И., Китаев Д.Б., Волков Ю.Ф. и др. // Радиохимия. 2001. Т. 43. № 3. С. 202.]https://doi.org/10.1023/A:1012844121536
Zubkova N.V., Kabalov Y.K., Orlova A.I. et al. // Crystallogr. Reports. 2003. V. 48. № 3. P. 401. [Зубкова Н.В., Кабалов Ю.К., Орлова А.И. и др. // Кристаллография. 2003. Т. 48. № 3. С. 445.]https://doi.org/10.1134/1.1578122
Kitaev D.B., Volkov Y.F., Orlova A.I. // Radiochemistry. 2004. V. 46. № 3. P. 211. [Китаев Д.Б., Волков Ю.Ф., Орлова А.И. // Радиохимия. 2004. Т. 46. № 3. С. 195.]https://doi.org/10.1023/B:RACH.0000031674.74780.a8
Orlova M.P., Kitaev D.B., Spiridonova M.L. et al. // Crystallogr. Reports. 2005. V. 50. № 6. P. 918. [Орлова М.П., Китаев Д.Б., Спиридонова М.Л. и др. // Кристаллография. 2005. Т. 50. № 6. С. 993.]https://doi.org/10.1134/1.2132396
Orlova A.I., Kitaev D.B., Kazantsev N.G. et al. // Radiochemistry. 2002. V. 44. № 4. P. 326. [Орлова А.И., Китаев Д.Б., Казанцев Н.Г. и др. // Радиохимия. 2002. Т. 44. № 4. С. 299.]https://doi.org/10.1023/A:1020656407183
Popa K., Bregiroux D., Konings R.J.M. et al. // J. Solid State Chem. 2007. V. 180. № 8. P. 2346. https://doi.org/10.1016/j.jssc.2007.06.006
Shekunova T.O., Istomin S.Y., Mironov A.V. et al. // Eur. J. Inorg. Chem. 2019. V. 2019. № 27. P. 3242. https://doi.org/10.1002/ejic.201801182
Salvadó M.A., Pertierra P., Bortun A.I. et al. // Inorg. Chem. 2008. V. 47. № 16. P. 7207. https://doi.org/10.1021/ic800818c
Matković B., Prodić B., Sljukić M. et al. // Croat. Chem. Acta. 1968. V. 40. P. 147. https://hrcak.srce.hr/208043
Matković B., Kojić-Prodić B., Šlijukić M. et al. // Inorg. Chim. Acta. 1970. V. 4. P. 571. https://doi.org/10.1016/S0020-1693(00)93352-8
Locock A.J. // Struct. Chem. Inorg. Actin. Compd / Eds. Krivovichev I.G. et al. Amsterdam: Elsevier, 2007. P. 217.
Guesdon A., Provost J., Raveau B. // J. Mater. Chem. 1999. V. 9. № 10. P. 2583. https://doi.org/10.1039/a904121d
Shannon R.D., Prewitt C.T. // Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem. 1969. V. 25. № 5. P. 925. https://doi.org/10.1107/s0567740869003220
Sidey V. // Acta Crystallogr., Sect. B: Struct. Sci. Cryst. Eng. Mater. 2016. V. 72. № 4. P. 626. https://doi.org/10.1107/S2052520616008064
Bamberger C.E., Begun G.M., Brynestad J. et al. // Radiochim. Acta. 1982. V. 31. № 1–2. P. 57. https://doi.org/10.1524/ract.1982.31.12.57
Botto I.L., Baran E.J. // ZAAC J. Inorg. Gen. Chem. 1977. V. 430. № 1. P. 283. https://doi.org/10.1002/zaac.19774300129
Dacheux N., Podor R., Brandel V. et al. // J. Nucl. Mater. 1998. V. 252. № 3. P. 179. https://doi.org/10.1016/S0022-3115(97)00337-1
Nazaraly M., Wallez G., Chanéac C. et al. // J. Phys. Chem. Solids. 2006. V. 67. P. 1075. https://doi.org/10.1016/j.jpcs.2006.01.028
Xu Y., Feng S., Pang W. et al. // Chem. Commun. 1996. № 11. P. 1305. https://doi.org/10.1039/CC9960001305
Xu Y., Feng S., Pang W. // Mater. Lett. 1996. V. 28. № 4–6. P. 499. https://doi.org/10.1016/0167-577X(96)00112-7
Salvado M.A., Pertierra P., Trobajo C. et al. // J. Am. Chem. Soc. 2007. V. 129. № 36. P. 10970. https://doi.org/10.1021/ja0710297
Bregiroux D., Popa K., Wallez G. // J. Solid State Chem. 2015. V. 230. P. 26. https://doi.org/10.1016/j.jssc.2015.06.010
Popa K., Konings R.J.M., Wiss T. et al. // J. Radioanal. Nucl. Chem. 2007. V. 273. № 3. P. 563. https://doi.org/10.1007/s10967-007-0910-x
Lai Y., Chang Y., Wong T. et al. // Inorg. Chem. 2013. V. 52. № 23. P. 13639. https://doi.org/10.1021/ic402208s
Xu Y.-H., Wang D., Feng S.-H. et al. // Chem. Res. Chin. Univ. 2000. V. 16. P. 287.
Bevara S., Achary S.N., Patwe S.J. et al. // Dalton Trans. 2016. V. 45. № 3. P. 980. https://doi.org/10.1039/c5dt03288a
Bevara S., Achary S.N., Patwe S.J. et al. // AIP Conf. Proc. 2016. V. 1731. P. 1. https://doi.org/10.1063/1.4948206
Bevara S., Mishra K.K., Patwe S.J. et al. // Inorg. Chem. 2017. V. 56. № 6. P. 3335. https://doi.org/10.1021/acs.inorgchem.6b02870
Bevara S., Rajeswari B., Patwe S.J. et al. // J. Alloys Compd. 2019. V. 783. P. 310. https://doi.org/10.1016/j.jallcom.2018.12.315
Yu N., Klepov V.V., Schlenz H. et al. // Cryst. Growth Des. 2017. V. 17. № 3. P. 1339. https://doi.org/10.1021/acs.cgd.6b01741
Ogorodnyk I.V., Zatovsky I.V., Baumer V.N. et al. // Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 2006. V. 62. № 12. P. 100. https://doi.org/10.1107/S0108270106044519
Майоров П.А., Асабина Е.А., Петьков В.И. и др. // Журн. неорган. химии. 2020. Т. 65. № 5. С. 660. [Mayorov P.A., Asabina E.A., Pet’kov V.I. et al. // Russ. J. Inorg. Chem. 2020. V. 65. № 5. P. 711. https://doi.org/10.1134/S0036023620050137]https://doi.org/10.31857/S0044457X2005013X
Clearfield A., Troup J.M. // J. Phys. Chem. 1973. V. 77. № 2. P. 243. https://doi.org/10.1021/j100621a022
Bohre A., Avasthi K., Pet’kov V.I. // J. Ind. Eng. Chem. 2017. V. 50. P. 1. https://doi.org/10.1016/j.jiec.2017.01.032
König K.-H., Eckstein G. // J. Inorg. Nucl. Chem. 1969. V. 31. № 4. P. 1179. https://doi.org/10.1016/0022-1902(69)80167-3
Lebedev V.N., Rudenko A.V. // Russ. J. Appl. Chem. 2002. V. 75. № 8. P. 1357. [Лебедев В.Н., Руденко А.В. // Журн. прикл. химии. 2002. Т. 75. № 8. С. 1384.]https://doi.org/10.1023/A:1020941817972
Kozlova T.O., Mironov A.V., Istomin S.Y. et al. // Chem. A. Eur. J. 2020. V. 26. № 53. P. 12188. https://doi.org/10.1002/chem.202002527
Hardwick T.J., Robertson E. // Can. J. Chem. 1951. V. 29. № 10. P. 818. https://doi.org/10.1139/v51-094
Lindgren O. // Acta Chem. Scand. 1977. V. 31a. P. 163. https://doi.org/10.3891/acta.chem.scand.31a-0163
Dacheux N., Clavier N., Wallez G. et al. // Solid State Sci. 2007. V. 9. № 7. P. 619. https://doi.org/10.1016/j.solidstatesciences.2007.04.015
Белова Л.Н., Горшков А.И., Иванова О.А. и др. // Записки Российского минералогического общества. 1984. Т. 113. № 3. С. 360.
Steciuk G., Ghazisaeed S., Kiefer B. et al. // RSC Adv. 2019. V. 9. № 34. P. 19657. https://doi.org/10.1039/c9ra03694f
Brandel V., Dacheux N. // J. Solid State Chem. 2004. V. 177. № 12. P. 4755. https://doi.org/10.1016/j.jssc.2004.08.008
Gebert W., Tillmanns E. // Acta Crystallogr. 1975. V. B31. № 6. P. 1768. https://doi.org/10.1107/S0567740875006115
Wallez G., Launay S., Souron J.P. et al. // Chem. Mater. 2003. V. 15. № 20. P. 3793. https://doi.org/10.1021/cm031058q
Черноруков Н.Г., Коршунов И.A., Жук M.И. // Журн. неорган. химии. 1983. Т. 28. № 7. С. 1656.
Wallez G., Souron J.-P., Quarton M. // Solid State Sci. 2006. V. 8. № 9. P. 1061. https://doi.org/10.1016/j.solidstatesciences.2006.02.054
Bénard-Rocherullé P., Louër M., Louër D. et al. // J. Solid State Chem. 1997. V. 132. № 2. P. 315. https://doi.org/10.1006/jssc.1997.7466
Sato T., Li R., Sato C. et al. // Phosphorus Res. Bull. 2007. V. 21. P. 44. https://doi.org/10.3363/prb.21.44
Bénard P., Brandel V., Dacheux N. et al. // Chem. Mater. 1996. V. 8. № 1. P. 181. https://doi.org/10.1021/cm950302d
Yu R., Wang D., Takei T. et al. // J. Solid State Chem. 2001. V. 157. № 1. P. 180. https://doi.org/10.1006/jssc.2000.9072
Yu R., Wang D., Ishiwata S. et al. // Chem. Lett. 2004. V. 33. № 4. P. 458. https://doi.org/10.1246/cl.2004.458
Brandel V., Dacheux N., Rousselle J. et al. // Comptes Rendus Chim. 2002. V. 5. № 8–9. P. 599. https://doi.org/10.1016/S1631-0748(02)01419-4
Felder J.B., Calder S., Zur Loye H.C. // Inorg. Chem. 2018. V. 57. № 15. P. 9286. https://doi.org/10.1021/acs.inorgchem.8b01284
Bray T.H., Sullens T.A., Shvareva T.Y. et al. // J. Solid State Chem. 2007. V. 180. № 1. P. 70. https://doi.org/10.1016/j.jssc.2006.09.019
Leblanc M., Maisonneuve V., Tressaud A. // Chem. Rev. 2015. V. 115. № 2. P. 1191. https://doi.org/10.1021/cr500173c
Brahim A., Mohamed Mongi F., Amor H. // Acta Crystallogr., Sect. E: Struct. Reports Online 2002. V. 58. № 11. P. I98. https://doi.org/10.1107/s1600536802018664
Liu J.Y., Wu D., Sun W.M. et al. // Dalton Trans. 2014. V. 43. № 48. P. 18066. https://doi.org/10.1039/c4dt02347a
Yu N., Klepov V.V., Modolo G. et al. // Inorg. Chem. 2014. V. 53. № 20. P. 11231. https://doi.org/10.1021/ic5018246
Rouse J., Weller M.T. // J. Chem. Soc., Dalton Trans. 2009. V. 44. № 46. P. 10330. https://doi.org/10.1039/b912262a
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии