

Журнал неорганической химии, 2021, T. 66, № 12, стр. 1748-1753
Влияние поправки на кулоновское самодействие 3d-электронов переходных металлов на электронные и магнитные свойства соединений CoGeN2, CrGeN2, MnSiN2 и MnGeN2
В. Б. Кольцов a, *, М. С. Михайлова a
a Московский институт электронной техники
124498 Москва, пл. Шокина, 1, Россия
* E-mail: koltsov_v_b@mail.ru
Поступила в редакцию 16.12.2020
После доработки 15.06.2021
Принята к публикации 16.06.2021
Аннотация
Электронная структура рассчитана для новых кристаллов CoGeN2, CrGeN2, MnSiN2 и MnGeN2 со структурой халькопирита в антиферромагнитной (АФМ) и ферромагнитной (ФМ) фазах. Установлено, что АФМ-состояние является энергетически выгодным для всех четырех материалов. Впервые изучены электронные свойства CoGeN2 и CrGeN2 с помощью двух обменно-корреляционных функционалов, а именно GGA-PBE и PBE0. Второй подход отличается от первого устранением ошибки кулоновского самодействия сильно коррелированных 3d-электронов. Например, без учета этой ошибки ширина запрещенной зоны в CoGeN2, MnSiN2 и MnGeN2 в АФМ-фазе составляет 0.11. 1.45 и 0.91 эВ, а с учетом – 1.46, 3.47 и 1.37 эВ соответственно. Соединение CoGeN2 без учета ошибки самодействия проявляет свойства металла для обоих ориентаций спина, а с ее учетом является прямозонным полупроводником с шириной запрещенной зоны 1.38 эВ для спина вверх и 1.77 эВ для спина вниз. Рассматриваемые кристаллы являются перспективными материалами для приложений магнитоэлектроники.
ВВЕДЕНИЕ
В настоящее время ведется поиск эффективных материалов для спиновой электроники. Один из подходов состоит в сплавлении известных и хорошо изученных полупроводниковых материалов с атомами переходных 3d-элементов. В частности, большое внимание уделяется исследованию полупроводников AIIIBV, легированных Mn, таких как GaMnAs, GaMnN, и кремния, легированного 3d-переходными элементами (Si : T, T = V, Cr, Mn, Fe, Co, Ni ) [1]. В работе [2] исследованы магнитные свойства сплавов InMnSb с целью поиска эффективных материалов спинтроники. Теоретические аспекты материалов, содержащих подсистему сильно коррелированных 3d-электронов, рассмотрены в работах [3–5] с привлечением модельных и априорных функционалов обменно-корреляционного потенциала. В работе [5] исследовано локальное обменное взаимодействие s-электронов валентной зоны с 3d-электронами Mn, причем обменный интеграл рассчитан с использованием точных атомных волновых функций. Температура Кюри фазового перехода из ферромагнитного (ФМ) в парамагнитное состояние определена через понижение полной энергии за счет спиновой поляризации в расчете на одну формульную единицу.
Поиск перспективных материалов для спинтроники ведется также путем исследования полусплавов Гейслера [6]. Инверсные сплавы Гейслера X2YZ (X = Cr; Y = Co, Ni; Z = Al, Ga, In, Si, Ge, Sn, Sb) выявили ферримагнитный порядок, обусловленный взаимодействием магнитных моментов атомов Cr–Cr, поэтому они являются перспективными кандидатами для применения в сканирующей туннельной микроскопии для исключения паразитного поля, индуцированного в случае наконечника из ФМ-материала [7].
Халькопириты с кристаллической структурой относятся к семейству материалов на основе цинковой обманки. Халькопириты, легированные Mn, такие как CdGeP2 [8], ZnSnAs2 [9] и ZnGeP2 [10], демонстрируют ФМ-упорядочение с температурой Кюри 320, 329 и 312 K соответственно.
Следует подчеркнуть, что в контексте спиновой электроники большое внимание уделяется спин-поляризованным полуметаллическим системам [6, 11]. Но сегодня можно говорить о новом направлении исследований, посвященном антиферромагнитной (АФМ) спинтронике [12–14]. Поэтому поиск перспективных материалов для использования в спинтронике является актуальной задачей современности. В настоящей работе исследовано электронное строение новых соединений со структурой халькопирита при помощи двух обменно-корреляционных функционалов энергии.
ТЕОРЕТИЧЕСКАЯ ЧАСТЬ
Все представленные здесь расчеты были выполнены на основе базиса проекционно-присоединенных волн (projector augmented waves, PAW) [15] с использованием программного пакета ABINIT [16]. Гибридный функционал обменно-корреляционной энергии рассчитывали в приближении PBE0 [17], которое состоит из двух слагаемых: полулокального вклада, основанного на обобщенном градиентном приближении (GGA-PBE) [18], и обменной энергии в приближении Хартри–Фока (ХФ, HF). Таким образом, обменно-корреляционная энергия в подходе PBE0 является суммой двух слагаемых, в которую энергия ХФ включается при помощи параметра смешивания ${{\alpha }}$. Параметр ${{\alpha }}$ = 0.25 рекомендуется использовать при расчетах электронной структуры материалов, содержащих d(f)-переходные элементы.
Применение функционалов $E_{{{\text{xc}}}}^{{{\text{PBE}}}}$ (${{\alpha }}$ = 0) и $E_{{{\text{xc}}}}^{{{\text{PBE}}0}}$ (${{\alpha }}$ = 0.25) дает возможность оценить влияние ошибки кулоновского самодействия (ОКС, self-interaction error, SIE) сильно коррелированных 3d-электронов на электронный энергетический спектр кристаллов, содержащих 3d-элементы Cr, Co и Mn. Применение гибридного функционала PBE0 привело к значительно лучшему согласию теоретических и экспериментальных значений кинетических коэффициентов A2B6 [19, 20].
Симметрия рассматриваемых здесь кристаллов описывается пр. гр. Pna21 (№ 33), а магнитная точечная группа для АФМ-фазы – m'm2'. Элементарная ячейка рассматриваемых здесь кристаллов ABN2 имеет четыре формульные единицы (Z = 4) и содержит 16 атомов. Базис элементарной ячейки определяется тройкой взаимно перпендикулярных векторов $\left( {{{{\mathbf{a}}}_{1}},{{{\mathbf{a}}}_{2}},{{{\mathbf{a}}}_{3}}} \right)$, модули которых равны $a,b,c$. Базис обратной решетки – это тройка взаимно перпендикулярных векторов $\left( {{{{\mathbf{b}}}_{1}},{{{\mathbf{b}}}_{2}},{{{\mathbf{b}}}_{3}}} \right)$, модули которых равны ${{2\pi } \mathord{\left/ {\vphantom {{2\pi } a}} \right. \kern-0em} a},{{2\pi } \mathord{\left/ {\vphantom {{2\pi } b}} \right. \kern-0em} b},{{2\pi } \mathord{\left/ {\vphantom {{2\pi } c}} \right. \kern-0em} c}$. Последние определяют положения точек высокой симметрии в первой зоне Бриллюэна, приведенные координаты которых равны $X\left( {{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2},0,0} \right),$ $\Gamma \left( {0,0,0} \right),$ $U\left( {{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2},0,{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}} \right),$ $Z\left( {0,0,{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}} \right),$ $S\left( {{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2},{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2},0} \right),$ $T\left( {0,{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2},{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}} \right),$ $R\left( {{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2},{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2},{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}} \right),$ $Y\left( {0,{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2},0} \right)$. Однако расчеты выполняются после умножения приведенных координат на $\pi $. Например, после умножения координаты точки R составляют $\left( {{\pi \mathord{\left/ {\vphantom {\pi 2}} \right. \kern-0em} 2},{\pi \mathord{\left/ {\vphantom {\pi 2}} \right. \kern-0em} 2},{\pi \mathord{\left/ {\vphantom {\pi 2}} \right. \kern-0em} 2}} \right)$. Именно эти точки зоны Бриллюэна изображены ниже на рисунках.
Магнитные свойства кристаллов рассчитывали в ФМ- и АФМ-фазах как с учетом ОКС, так и без учета последней. Оба подхода оперируют плотностью электронов с противоположными спинами ${{s}_{ \uparrow }}$ и ${{s}_{ \downarrow }},$ т.е. в результате самосогласованного вычисления мы получаем разность ($n\left( {{{s}_{ \uparrow }},{\mathbf{r}}} \right) - n\left( {{{s}_{ \downarrow }},{\mathbf{r}}} \right)$), интеграл которой после умножения на магнетон Бора дает магнитный момент элементарной ячейки. Этот подход в программе ABINIT реализуется выбором режима с двумя спиновыми поляризациями для волновой функции и плотности электронов. Тогда подход без учета ОКС ($\alpha $ = 0) вообще не содержит внешних параметров, а учитывая ОКС, мы имеем дело только с единственным параметром – $\alpha $ = 0.25. Это полный спин-поляризованный подход, в котором нет необходимости использовать эмпирические параметры U и J.
Элементарные ячейки рассматриваемых кристаллов содержат четыре атома переходного 3d-металла, которые являются попарно симметрично-эквивалентными. Например, в кристалле CoGeN2 эквивалентными являются атомы (Co1, Co3) и (Co2, Co4). Это сильно упрощает реализацию режима расчета в ФМ- или АФМ-фазе. Для режима ФМ следует задать начальные максимальные значения спинов атомов Co как (3, 3, 3, 3), для АФМ – (3, –3, 3, –3). Удачный выбор начального приближения приводит к ускорению процесса сходимости самосогласованных расчетов.
Базис плоских волн определяется максимальной кинетической энергией 60 Ry с сеткой 54 × × 64 × 50 для волновой функции, более плотная сетка – максимальной энергией 240 Ry с сеткой 108 × 128 × 100 для расчета электронной плотности и потенциала.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Энергетический спектр электронов кристалла CoGeN2 представлен на рис. 1. За реперный уровень энергии электронов принята энергия Ферми. Последняя расположена в запрещенной зоне, как в классическом полупроводнике. Нами установлено, что верхняя часть валентной зоны образована p-состояниями атомов Ge и N. Эти состояния доминируют и в нижней части зоны проводимости.
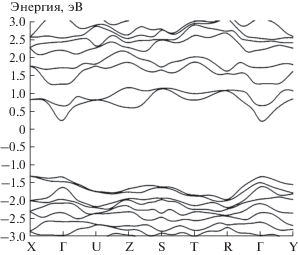
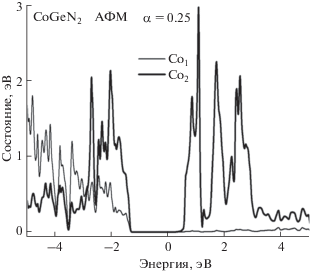
Парциальные плотности PDOS двух различных атомов Co показаны на рис. 2. Кривые демонстрируют значительную симметричную неэквивалентность двух атомов кобальта в элементарной ячейке. Как видно из рис. 2, 3d-электроны атомов Co движутся в узких энергетических зонах, а значения PDOS большие. Без учета ОКС 3d-электронов (α = 0) вычисления дают непрямую запрещенную зону в направлении Γ–Z, равную 0.11 эВ. Включение в расчет ОКС (α = 0.25) приводит к непрямозонному полупроводнику с запрещенной зоной ${{\varepsilon }_{g}}$ = 1.46 эВ в направлении Γ–X. Поскольку 3d-электроны атомов Co представляют собой сильно коррелированную подсистему фермионов, целесообразно применение гибридного функционала PBE0. Рассчитанные без учета ОКС (α = 0) магнитные моменты атомов Co1, Co2, Co3 и Co4 имеют значения 2.23, –2.23, 2.23 и –2.23 μB, а с учетом поправок (α = 0.25) – 2.64, –2.64, 2.64 и –2.64 μB соответственно. Полный магнитный момент элементарной ячейки равен нулю.
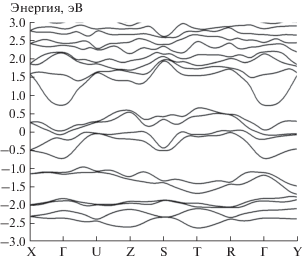
Кривые дисперсии электронов валентной зоны, полученные в кристалле CrGeN2 без учета ОКС 3d-электронов, пересекают уровень Ферми. Ближайшая малая щель расположена на ~0.6 эВ выше уровня Ферми, который погружен в валентную зону.
Найденные без учета ОКС (α = 0) магнитные моменты атомов Cr1, Cr2, Cr3 и Cr4 имеют значения 3.02, –3.02, 3.02 и –3.02 μB, а с учетом ОКС (α = 0.25) – 3.19, –3.19, 3.19 и –3.19 μB соответственно. Полный магнитный момент элементарной ячейки равен нулю.
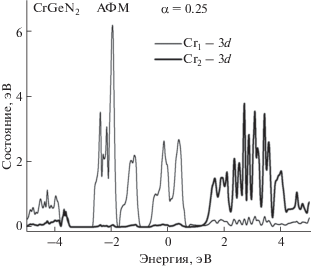
Учет ОКС (α = 0.25) не приводит к каким-либо заметным изменениям зонных кривых, приведенных на рис. 3. Уровень Ферми погружен в заполненные состояния валентной зоны. Результаты расчета PDOS 3d-электронов (рис. 4), реализованные с учетом ОКС, показывают значительные изменения в локализации $n\left( E \right)$ Cr.
Электронные энергетические зоны кристалла MnSiN2, найденные с пренебрежением ОКС (рис. 5), приводят к локализации уровня Ферми внутри запрещенной зоны, как в полупроводнике. Кристалл имеет непрямую запрещенную зону в направлении RT–X, равную 1.45 эВ.
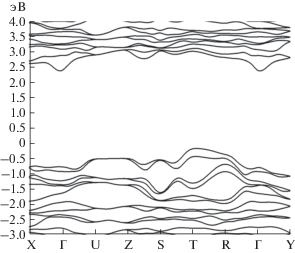
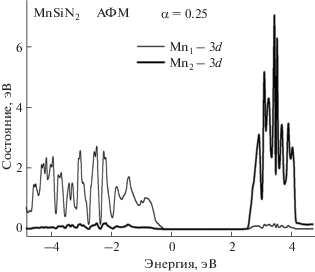
Магнитные моменты атомов Mn1, Mn2, Mn3 и Mn4 в элементарной ячейке, найденные без учета ОКС (α = 0), имеют значения 3.97, –3.97, 3.97 и ‒3.97 μB соответственно, а с учетом ОКС они равны 4.25, –4.25, 4.25 и –4.25 μB. Полный магнитный момент элементарной ячейки равен нулю. Учет ОКС приводит к увеличению ширины запрещенной зоны материала MnSiN2, а также к значительному изменению PDOS 3d-электронов атомов Mn в сравнении с результатами, полученными с пренебрежением ОКС (рис. 6).
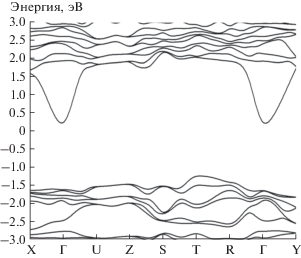
Игнорируя поправку на ОКС (α = 0), мы установили, что в кристалле MnGeN2 уровень Ферми немного смещен в сторону зоны проводимости. Кристалл имеет непрямую запрещенную зону в направлении T–Γ, равную 0.91 эВ.
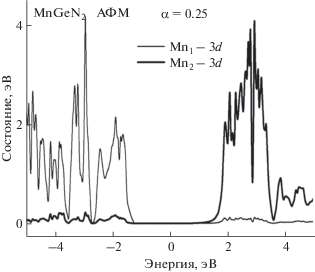
Без учета поправки на ОКС рассчитанные магнитные моменты атомов Mn1, Mn2, Mn3 и Mn4 имеют значения 3.97, –3.97, 3.97 и –3.97 μB, однако учет ОКС приводит к значениям 4.24, –4.24, 4.24 и –4.24 μB соответственно. Суммарный магнитный момент элементарной ячейки равен нулю. Энергетические зоны электронов в кристалле MnGeN2, полученные с поправками на ОКС (α = = 0.25), представлены на рис. 7. Хорошо видно, что уровень Ферми находится внутри запрещенной зоны и смещен в сторону зоны проводимости. Кристалл имеет непрямую запрещенную зону в направлении T–Γ, равную 1.37 эВ.
Кривые PDOS атомов Mn представлены на рис. 8. Они рассчитаны с учетом ОКС для подсистемы 3d-электронов. Кривые, соответствующие неэквивалентным атомам Mn в элементарной ячейке, показывают распределение 3d-электронов по энергии. Кривые PDOS, представленные на рис. 8, заполняют более широкие энергетические интервалы как в валентной зоне, так и в зоне проводимости по сравнению с таковыми, полученными без учета поправок на ОКС.
Соединения MnGeN2 и MnSiN2 ранее исследовались на основе PAW в рамках обменно-корреляционного функционала GGA, но только в ФМ-фазе [21]. Магнитные моменты на элементарную ячейку, найденные здесь в ФМ-фазе для кристаллов MnGeN2 и MnSiN2, равны 20.0 μB. Эти значения очень хорошо согласуются с таковыми, рассчитанными на формульную единицу для обоих кристаллов [21], которые составляют 5.0 μB.
Недавние публикации, посвященные электронному строению соединений HgCX2 [22, 23], свидетельствуют о возрастании интереса к материалам, изоструктурным рассмотренным в настоящей работе.
ЗАКЛЮЧЕНИЕ
Электронные свойства кристаллических халькопиритов CoGeN2, CrGeN2, MnSiN2 и MnGeN2 были изучены в АФМ- и ФМ-фазах. Результаты расчетов показали, что учет ошибки кулоновского самодействия приводит к значительному изменению энергии 3d-электронов переходных металлов в кристалле. Результаты, полученные нами в АФМ- и ФМ-фазах, показывают значительное влияние учета ошибки кулоновского самодействия на значения ширины запрещенной зоны кристаллов. Разность полных энергий EФМ–EАФМ составляет 1.70, 0.41, 1.54 и 1.33 эВ для материалов CoGeN2, CrGeN2, MnSiN2 и MnGeN2 соответственно. Это значения указывают на более устойчивое состояние АФМ по сравнению с ФМ. Рассмотренные кристаллы являются перспективными материалами для приложений магнитоэлектроники.
Список литературы
Zhang Z.Z., Partoens B., Kai Chang et al. // Phys. Rev. B. 2008. V. 77. № 15. P. 155201. https://doi.org/10.1103/PhysRevB.77.155201
Marenkin S., Kochura A., Fedorchenko I. et al. // Inorg. Mater. 2016. V. 52. № 3. P. 268. https://doi.org/10.1134/S0020168516030110
Yarzhemsky V.G., Murashov S.V., Izotov A.D. // Inorg. Mater. 2016. V. 52. P. 89. https://doi.org/10.1134/S0020168516020175
Yarzhemsky V.G., Murashov S.V., Izotov A.D. // Inorg. Mater. 2017. V. 53. P. 1131. https://doi.org/10.1134/S0020168517110176
Yarzhemsky V.G., Murashov S.V., Izotov A.D. // Inorg. Mater. 2017. V. 55. P. 1. https://doi.org/10.1134/S0020168519010187
Graf T., Felser C., Parkin S.S.P. // Prog. Solid State Chem. 2011. V. 39. № 1. P. 1. https://doi.org/10.1016/j.progsolidstchem.2011.02.001
Susanta K. Mohanta, Yongxue Tao, Xiaoyan Yan et al. // J. Magn. Magn. Mater. 2017. V. 430. № 1. P. 65. https://doi.org/10.1016/j.jmmm.2017.01.055
Medvedkin G.A., Ishibashi T., Nishi T. et al. // Jpn. J. Appl. Phys. 2000. V. 39. P. L949. https://doi.org/10.1007/s10948-005-2154-8
Choi S., Cha G.B., Hong S. et al. // Solid State Commun. 2002. V. 122. № 3–4. P. 165. https://doi.org/10.1016/S0038-1098(02)00094-7
Cho S., Choi S., Cha G.B. et al. // Phys. Rev. Lett. 2002. V. 88. № 25. P. 257203. https://doi.org/10.1103/PhysRevLett.88.257203
Pogorily A., Ryabchenko S., Tovstolytkin A. // Ukr. J. Phys. Rev. 2010. V. 6. № 1. P. 37.
Gomonay E.V., Loktev V.M. // Low Temp. Phys. 2014. V. 40. № 1. P. 22. https://doi.org/10.1063/1.4862467
Kravchuk V.P., Gomonay O., Sheka D.D. et al. // Phys. Rev. B. 2019. V. 99. № 18. P. 184429. https://doi.org/10.1103/PhysRevB.99.184429
Grytsiuk S., Hanke J.-P., Hoffmann M. et al. // Nat. Commun. 2020. V. 11. № 1. P. 1. https://doi.org/10.1038/s41467-019-14030-3
Blöchl P.E. // Phys. Rev. B. 1994. V. 50. № 24. P. 17953. https://doi.org/10.1103/PhysRevB.50.17953
Gonze X., Jollet F., Abreu Araujo F. et al. // Comput. Phys. Commun. 2016. V. 205. № 8. P. 106. https://doi.org/10.1016/j.cpc.2016.04.003
Novák P., Kuneš J., Chaput L. et al. // Phys. Status Solidi B. 2006. V. 243. № 3. P. 563. https://doi.org/10.1002/pssb.200541371
Perdew J.P., Burke K., Ernzerhof M. // Phys. Rev. Lett. 1996. V. 77. № 18. P. 3865. https://doi.org/10.1103/PhysRevLett.77.3865
Malyk O.P., Syrotyuk S.V. // Comput. Mater. Sci. 2017. V. 139. № 11. P. 387. https://doi.org/10.1016/j.commatsci.2017.07.039
Malyk O.P., Syrotyuk S.V. // J. Electron. Mater. 2018. V. 47. № 8. P. 4212. https://doi.org/10.1007/s11664-018-6068-1
Naveh D., Kronik L. // Phys. Status Solidi B. 2006. V. 243. № 9. P. 2159. https://doi.org/10.1002/pssb.200666806
Basalaev Yu.M., Basalaeva M.Yu. // J. Struct. Chem. 2020. V. 61. P. 1007. https://doi.org/10.1134/S0022476620070021
Basalaev Yu.M., Basalaeva M.Yu., Duginova E.B. et al. // J. Struct. Chem. 2020. V. 61. P. 1839. https://doi.org/10.1134/S002247662012001X
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии