

Журнал неорганической химии, 2021, T. 66, № 12, стр. 1723-1731
Комплексообразующие свойства 2-окси-5-этилфенилфосфоновой кислоты (Н3L). Кристаллическая структура и анальгетическая активность [Cu(H2L)2(Н2О)2]
И. С. Иванова a, Г. С. Цебрикова b, *, Ю. И. Рогачева c, А. Б. Илюхин a, В. П. Соловьев b, Е. Н. Пятова a, В. Е. Баулин c
a Институт общей и неорганической химии им. Н.С. Курнакова РАН
119991 Москва, Ленинский пр-т, 31, Россия
b Институт физической химии и электрохимии им. А.Н. Фрумкина РАН
119991 Москва, Ленинский пр-т, 31, корп. 4, Россия
c Институт физиологически активных веществ РАН
142432 Черноголовка, Московской обл., Северный пр-д, 1, Россия
* E-mail: tsebrikova@yandex.ru
Поступила в редакцию 24.04.2021
После доработки 31.05.2021
Принята к публикации 01.06.2021
Аннотация
Взаимодействием меди(II) c 2-окси-5-этилфенилфосфоновой кислотой (Н3L) синтезирован комплекс [Cu(H2L)2(Н2О)2] и методом РСА изучена его структура. Методом потенциометрического титрования определены константы протонирования кислоты Н3L и константы устойчивости ее комплексов с Cu2+ в воде. Установлено, что комплекс [Cu(H2L)2(Н2О)2] обладает малой токсичностью и высокой анальгетической активностью.
ВВЕДЕНИЕ
Известно [1–7], что фосфорорганические соединения играют большую роль в органическом синтезе, катализе и биохимии, а фосфорсодержащие фенолы являются необходимыми компонентами в синтезе более сложных структур [8, 9]. Методы синтеза различных фосфорилсодержащих фенолов недавно были проанализированы авторами [10–12]. В работе [11] описан синтез 2-оксифенилфосфонистой (L1), 2-оксифенилфосфоновой (L2) и 2-окси-5-этилфенилфосфоновой (Н3L) кислот, которые относятся к классу 2-фосфорилфенолов. Эти соединения являются структурными аналогами салициловой кислоты (L3), в которой C(O)OH-группа заменена на группу P(O)(OH)2. Кислота L3 является хорошо известным органическим лигандом, производные которого обладают противовоспалительным, жаропонижающим и обезболивающим действием [13] и широко используются в современной фармацевтике [14]. Известно, что комплексы органических лигандов с катионами металлов часто проявляют более высокую биологическую активность и менее токсичны, чем исходные лиганды [15–18]. Так, комплексы меди с органическими лигандами обладают широким спектром биологической активности (противоопухолевой, антимикробной, противовоспалительной и др.) [19–26] и представляют интерес в качестве компонентов радиофармпрепаратов [27–30]. Однако данных о строении и свойствах координационных соединений катионов металлов с 2-фосфорилфенолами (фосфорильными аналогами салициловой кислоты) известно мало. Комплексы 2-фосфорилфенолов c некоторыми лантанидами(III) описаны в работе [31], в [32] представлены кристаллические структуры некоторых комплексов меди(II) с фосфорилфенолами.
В продолжение изучения [11] физико-химических, биологических свойств и строения фосфорильных аналогов салициловой кислоты в настоящей работе синтезирован комплекс 2-окси-5-этилфенилфосфоновой кислоты (Н3L) с катионом меди(II) – [Cu(H2L)2(Н2О)2]. Состав комплекса подтвержден методами элементного анализа, ИК-спектроскопии и термогравиметрии. Определены константы протонирования кислоты Н3L и константы устойчивости ее комплексов с Cu2+ в воде. Методом РСА установлена кристаллическая структура комплекса [Cu(H2L)2(Н2О)2]. Проведена оценка острой токсичности и анальгетической активности комплекса.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Синтез 2-окси-5-этилфенилфосфоновой кислоты (Н3L) описан в [11]. Комплекс [Cu(H2L)2(Н2О)2] получали взаимодействием водных растворов Н3L и Cu(ClO4)2 · 6H2O при эквимолярном соотношении исходных компонентов. Смесь растворов ярко-голубого цвета на непродолжительное время оставляли на воздухе. Выпавший светло-голубой кристаллический осадок отфильтровывали, промывали небольшим количеством воды и сушили на воздухе при комнатной температуре. Варьирование соотношения реагентов и проведение реакции в присутствии 1 экв NaOH не влияет на выход и состав образующегося осадка. Комплекс малорастворим в воде и малополярных органических растворителях, но хорошо растворим в этаноле, ДМФА и ДМСО. Кристаллы для РСА получены в результате медленной кристаллизации водного раствора.
Элементный анализ проводили на С,Н,N-анализаторе (Carlo Erba Strumentazione, Italy) и атомно-эмиссионном спектрометре с индуктивно связанной плазмой IRIS Advantage (“Thermo Jarrell Ash”, США).
ИК-спектры поглощения записывали в диапазоне 4000–550 см–1 методом НПВО на спектрометре Nexsus Nicolete.
Термогравиметрические исследования проводили на дериватографе SDT Q600 в диапазоне температур от 20 до 600°C при скорости нагревания 4 град/мин в токе аргона.
Константы протонирования кислоты H3L и константы устойчивости ее комплексов с перхлоратом меди(II) определяли методом потенциометрического титрования. Методика титрования с использованием потенциометра OP-300 Radelkis изложена в [33]. Для изучения комплексообразования использовали гексагидрат перхлората меди(II) марки “х. ч.”.
Четыре титрования Н3L, выполненные стандартным 0.1 М раствором NaOH при температуре 298 ± 0.1 K и ионной силе I = 0.1 M KCl, включали от 41 до 54 точек. Исходная аналитическая концентрация Н3L составляла 0.62, 0.80, 0.99 и 1.57 ммоль/л. Титрование проводили в интервале pH от 3.0 до 11.2. Константы протонирования Н3L определены с помощью программы CHEMEQUI с использованием трех алгоритмов [34, 35], позволяющих значительно повысить надежность оценок констант протонирования. Расчеты констант выполнены как для каждого эксперимента, так и для объединенных четырех экспериментов, обеспечивая наименьшие погрешности в оценке констант. В качестве критериев согласия предполагаемого набора равновесных реакций в растворе с экспериментальными данными использовали R-фактор Гамильтона (HRF) и коэффициент детерминации ($R_{{{\text{det}}}}^{{\text{2}}}$) [33]. В зависимости от эксперимента и алгоритма программы фактор HRF варьировали от 0.26 до 1.28%, а коэффициент $R_{{{\text{det}}}}^{{\text{2}}}$ – от 0.999 до 1.0. Всего выполнено 18 оценок констант, по которым вычислены их средние значения, за исключением, согласно правилу Томсона [36], резко отклоняющихся величин.
Титрования растворов Н3L с Cu(ClO4)2 ⋅ 6H2O, выполненные в аналогичных условиях, включали от 49 до 50 точек. Исходные аналитические концентрации Н3L и Cu(ClO4)2 ⋅ 6H2O в трех титрованиях составляли 0.76 и 0.36; 0.69 и 0.34; 0.41 и 0.21 ммоль/л. Титрование проводили в интервале pH от 3.3 до 10.9. Медь(II) образует в воде устойчивые гидроксиды [37], поэтому оценки констант комплексообразования Cu2+ с изучаемыми кислотами были выполнены как с учетом реакций гидролиза меди(II), так и без их учета. В расчетах использовали следующие константы устойчивости (lgβn) гидроксокомплексов в воде: –6.29 и –13.10 соответственно для равновесий Cu2+ + nH2O = = Cu2+(OH–)n + nH+, n = 1, 2 [37]. Оба подхода дали близкие оценки констант комплексообразования с пересекающимися интервалами их стандартных отклонений. Константы комплексообразования Н3L с Cu(ClO4)2 · 6H2O были оценены с помощью программы CHEMEQUI [34, 35] с использованием четырех алгоритмов: EQ, Simplex, Monte-Carlo и Genetic Algorithm [38]. В этих экспериментах фактор HRF варьировался от 0.88 до 1.68%, а $R_{{det}}^{2}$ – от 0.996 до 0.999. По трем титрованиям и четырем алгоритмам выполнено 12 оценок констант, по которым вычислены их средние значения, за исключением (по правилу Томсона) резко отклоняющихся величин [36]. В расчетах констант комплексообразования Cu2+ c протонированными формами HnL(3 –n)– (n = 0, 1, 2) константы диссоциации кислоты Н3L не варьировали, они взяты как ранее оцененные в предыдущих титрованиях Н3L. Оценки констант комплексообразования могут быть несколько смещены вследствие образования осадка – помутнения раствора при pH > 6, что свидетельствует о малой растворимости комплексов. Смещение оценок можно считать незначительным, поскольку при варьировании (оптимизации, что возможно с помощью программы CHEMEQUI) аналитической концентрации как лиганда, так и лиганда и меди(II) по результатам титрований были получены достаточно согласованные величины.
РСА. Экспериментальные данные для соединения [Cu(H2L)2(H2O)2] (I) получены на дифрактометре Bruker SMART APEX3 (λ(MoKα), графитовый монохроматор) [39], ЦКП ИОНХ РАН. Поглощение учтено полуэмпирическим методом по эквивалентам (программа SADABS [40]). Структура определена комбинацией прямого метода и синтезов Фурье. Кристалл оказался псевдомероэдрическим двойником – матрица 0/–1/0/0/0/–1/1/0/1 преобразует моноклинную ячейку в псевдоромбическую (a = 4.832, b = = 12.704, c = 33.232 Å, α = 90.03°, β = 90.00°, γ = = 90.00°), соотношение доменов равно 0.72 : 0.28. Метильный фрагмент лиганда разупорядочен по двум позициям в соотношении 1 : 1. Структура уточнена полноматричным анизотропно-изотропным (разупорядоченный атом C) МНК. Атомы водорода частично локализованы из разностного синтеза Фурье, частично рассчитаны из геометрических соображений. Все расчеты выполнены по программам SHELXS и SHELXL [41].
Экспериментальные данные для структуры I депонированы в Кембриджском банке структурных данных (CCDC № 2077739; deposit@ccdc.cam.ac.uk или http://www.ccdc.cam.ac.uk). Основные структурные данные приведены в табл. 1, геометрия водородных связей – в табл. 2.
Таблица 1.
Основные структурные данные и результаты уточнения структуры I
| Параметр | Значение |
|---|---|
| Соединение | I |
| T, K | 150(2) |
| Сингония | Моноклинная |
| Пр. гр. | С2/c |
| a, Å | 35.570(2) |
| b, Å | 4.8324(3) |
| c, Å | 12.7041(7) |
| β, град | 110.891(2) |
| V, Å3 | 2040.1(2) |
| Z | 4 |
| ρвыч, г/см3 | 1.634 |
| µ, мм–1 | 1.279 |
| Размер кристалла, мм | 0.30 × 0.20 × 0.12 |
| интервал θ, град | 2.452, 30.074 |
| Интервал индексов | –50 ≤ h ≤ 50 –6 ≤ k ≤ 6 –17 ≤ l ≤ 17 |
| Собранных отражений | 10739 |
| Независимых отражений (Rint) | 2978, 0.0940 |
| Полнота до θ = 25.242°, % | 99.9% |
| Max, min пропускание | 0.746, 0.5479 |
| Ограничения/ параметры | 0/135 |
| GООF | 1.053 |
| R1, wR2 (I > 2σ(I)) | 0.0644, 0.1658 |
| R1, wR2 (весь массив) | 0.0675, 0.1685 |
| Δρmax/Δρmin, е Å–3 | 1.787, –0.714 |
Таблица 2.
Геометрические параметры водородных связей в структуре I
| D–H…A | D–H, Å | H⋅⋅⋅A, Å | D⋅⋅⋅A, Å | ∠(DHA), град |
|---|---|---|---|---|
| O(3)–H(1)⋅⋅⋅O(2) (x, y + 1, z) | 0.90 | 1.60 | 2.498(4) | 175 |
| O(4)–H(2)⋅⋅⋅O(3) (x, –y + 1, z – 1/2) | 0.82 | 2.02 | 2.835(5) | 178 |
| O(5)–H(3)⋅⋅⋅O(1) (x, y – 1, z) | 0.90 | 1.82 | 2.719(5) | 180 |
| O(5)–H(4)⋅⋅⋅O(2) (–x + 1, y, –z + 3/2) | 0.90 | 1.92 | 2.722(5) | 148 |
Биологические испытания. Исследование острой токсичности H3L и комплекса [Cu(H2L)2(H2O)2] проводили путем однократного внутрижелудочного или внутрибрюшинного введения водного/водно-крахмального раствора мышам CD1 обоего пола массой 21–24 г. Наблюдение проводили в течение 14 сут. Значения ЛД50 рассчитывали по методу Литчфилда и Уилкоксона [42].
Анальгетическую активность H3L и [Cu(H2L)2(H2O)2] исследовали на белых беспородных мышах обоих полов массой 18–24 г на модели корчей, вызванных внутрижелудочным введением 0.25 мл 0.75%-ного раствора уксусной кислоты на 10.00 г веса животного. Исследуемые вещества вводили однократно в виде водного/водно-крахмального раствора в соотношении 0.10 мл на 100.00 г веса мыши за 1 ч до введения уксусной кислоты. Количество корчей подсчитывали в течение 15 мин. Каждую дозу исследовали на шести животных. Контрольным животным вводили соответствующий объем растворителя (вода), в случае комплекса добавляли крахмал. Активность оценивали по величине ЕД50, т.е. дозе, вызывающей 50% эффекта (уменьшения количества корч по сравнению с контролем). В качестве препарата сравнения использовали анальгин (ООО “ГРОТЕКС”, Россия).
Работа выполнена с соблюдением всех применимых международных, национальных и институциональных руководящих принципов по уходу и использованию животных.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
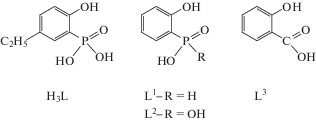
Структура I образована центросимметричными комплексами [Cu(H2L)2(H2O)2] (рис. 1а). Координационное окружение 4 + 2 обычно для Cu2+. Короткая водородная связь (ВС) O(3)–H…O(2) между некоординированными атомами O фосфоновых фрагментов объединяет комплексы в 1D-цепочку, параллельную оси b, совместное действие четырех ВС приводит к образованию 2D-структуры (слои перпендикулярны оси a), т.е. в структуре I присутствуют гидрофильные и гидрофобные области (рис. 1б). Отсутствие значимых вторичных взаимодействий в гидрофобной области приводит к разупорядоченности этильного фрагмента H2L.
Рис. 1.
Строение комплекса [Cu(H2L)2(H2O)2] в структуре I (Cu(1)–O(1) 1.967(3), Cu(1)–O(4) 2.448(3), Cu(1)–O(5) 1.949(3) Å) (а) и проекция структуры I вдоль оси b (б).
Известно, что в результате комплексообразования наибольшие изменения в ИК-спектрах испытывают частоты валентных колебаний донорных групп, принимающих участие в образовании координационных и водородных связей.
Отнесение некоторых колебательных частот донорных групп в ИК-спектрах Н3L и [Cu(H2L)2(H2O)2], позволяющих судить о координации лиганда, проведено с учетом совокупных спектральных и структурных исследований фосфорилподандов – производных бис-фосфоновых кислот, их комплексов [43–46] и ортозамещенных фенолов [47, 48], выполненных ранее.
Известно, что молекулы такого рода соединений, как кислота H3L, в свободном состоянии ассоциированы за счет образования ВС, в результате в ИК-спектрах полосы, обусловленные колебаниями донорных групп, могут быть существенно смещены в низкочастотную область [49]. В ИК-спектре H3L валентные колебания фенольной OH-группы ν(OH)Ph проявляются в виде широкой полосы средней интенсивности с максимумом около 2971 см–1 (~3600 см–1 в спектре свободного фенола). Полосы валентных колебаний фосфонатных OH-групп ν(OH)P лежат ниже, это малоинтенсивная широкая асимметричная полоса с максимумом при ~2690 см–1 и полоса около 2290 см–1. Существенное смещение в низкочастотную область и уширение полос валентных колебаний всех групп OH свидетельствуют об их участии в образовании водородных связей.
В ИК-спектре H3L около 1230 см–1 присутствует полоса средней интенсивности, которая, в соответствии со спектральными исследованиями [43–46], относится к ν(P=O). Полосу средней интенсивности при 1270 см–1 можно отнести к ν(Ph–O) фенольной группы [44, 49, 50].
Полосы деформационных колебаний фосфонового фрагмента δ(POH) должны лежать около 1000 см–1 [49]. Действительно, в ИК-спектре H3L в этой области наблюдаются две интенсивные полосы: дублетная при 991, 979 см–1 и интенсивная полоса при 949 см–1.
Комплексообразование приводит к незначительному понижению ν(P=O) в ИК-спектре [Cu(H2L)2(H2O)2] по сравнению со спектром свободного H3L (1230 → 1225 см–1). Похожая ситуация, когда фосфорильные атомы кислорода не участвуют в координации катиона меди, а изменение частоты ν(P=O) происходит только за счет образования водородных связей, наблюдалась в случае комплекса меди, описанного в [44]. Частота ν(Ph–O) в спектре комплекса повышается до 1284 см–1. В области деформационных колебаний фосфонового фрагмента в спектре комплекса появляется новая интенсивная полоса δ(POH) около 1017 см–1 вместо дублетной полосы (991, 979 см–1) в спектре H3L.
В ИК-спектре [Cu(H2L)2(H2O)2] также появляется новая широкая полоса с двумя максимумами около 3314 и 3201 см–1 вместо полосы при 2971 см–1 в спектре свободного H3L. Полоса при 3314 см–1 обусловлена, по-видимому, колебаниями ν(H2O) координированных молекул воды; колебания δ(H2O) проявляются около 1700 см–1 в виде широкой полосы средней интенсивности. Полоса при 3201 см–1 относится, на наш взгляд, к ν(OH)Ph, что значительно выше, чем в спектре свободного H3L. Полосы, обусловленные валентными колебаниями OH-групп фосфонового фрагмена ν(OH)P, участвующих в образовании водородных связей, смещаются в низкочастотную область: 2690 → 2570 и 2290 → 2273 см–1.
Термогравиметрические исследования [Cu(H2L)2(H2O)2] показали, что при нагревании комплекса при температуре ~124°С происходит удаление двух молекул воды, что составляет 7.88% от общей массы вещества (рассчитано 7.17%), которое на кривой ДТА сопровождается соответствующим эндотермическим эффектом. Столь высокое значение температуры указывает на внутрисферный характер молекул воды. Дальнейшее повышение температуры приводит к постепенному разложению комплекса, а на кривой ДТА появляются три эндотермических эффекта при 219, 284 и 333°С.
Методом потенциометрического титрования были определены константы протонирования кислоты H3L (табл. 3). Ранее для 2-оксифенилфосфоновой кислоты (L2) константы протонирования фосфоновой группы были определены методом потенциометрии, а константа протонирования фенольной группы – спектрофотометрическим методом [51]. Значения вторых констант lgK2 этих кислот близки: 6.36 ± 0.37 (H3L) и 6.46 (L2 [51]); значения же первой и третьей констант различаются значительно. Более низкая кислотность H3L (lgK1= 3.20 ± 0.74) по сравнению с L2 (lgK1= 1.66 [51]) обусловлена, по-видимому, присутствием донорного этильного заместителя, который затрудняет ионизацию фосфоновой группы. Кроме того, константа lgK1 была оценена со значительной величиной стандартного отклонения в связи с тем, что полную константу lgβ3 = 21.14 ± 0.69 (табл. 3) не удалось определить с достаточной точностью. Более низкая кислотность фенольной группы кислоты H3L (lgK3= 11.58 ± 0.24) по сравнению с величинами lgK3, равными 10.03 и 10.56 [51] соответственно для 3- и 4-оксифенилфосфоновых кислот, указывает на внутримолекулярную водородную связь кислоты H3L. Наличие этой связи в молекуле H3L подтверждено данными ИК-спектроскопии. Значение lgK3= 15.40 [51] для L2 представляется завышенным: маловероятно, чтобы в воде внутримолекулярная водородная связь изменила значение K3 на пять порядков по сравнению с K3 3- и 4-оксифенилфосфоновых кислот.
Таблица 3.
Ступенчатые и полные константы протонирования кислоты H3L в воде при температуре 298 K и ионной силе 0.1 M KCl a
| i | Равновесие | lgKi ± sdб | Равновесие | lgβi ± sdв |
|---|---|---|---|---|
| 1 | H2L + H = H3L | 3.20 ± 0.74 | L + H = HL | 11.58 ± 0.24 |
| 2 | HL + H = H2L | 6.36 ± 0.37 | L + 2H = H2L | 17.94 ± 0.28 |
| 3 | L + H = HL | 11.58 ± 0.24 | L + 3H = H3L | 21.14 ± 0.69 |
a Для простоты представления равновесий заряды химических форм не указаны: вместо H+ + L3– = HL2– приведено H + L = = HL и т.д. б lgKi и sd – ступенчатые константы равновесий и их стандартные отклонения, вычисленные по результатам нескольких титрований и расчетов (см. экспериментальную часть) c использованием закона сложения случайных ошибок и стандартных отклонений для полных констант равновесий lgβi. в lgβi и sd – полные константы равновесий и их стандартные отклонения, рассчитанные с помощью программы CHEMEQUI.
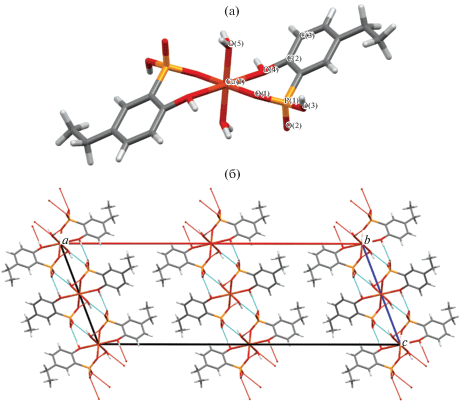
Диаграмма распределения протонированных форм HnL(3– n)– (n = 0, 1, 2, 3) кислоты H3L в зависимости от pH представлена на рис. 2. При физиологическом значении pH 7.4 в воде преобладает анион HL2– при миллимолярных концентрациях кислоты. В интервале рН от 3 до 5.5 кислота находятся преимущественно в форме аниона H2L–.
Рис. 2.
Распределение химических форм кислоты H3L в зависимости от pH в воде при 298 K, ионной силе 0.1 М и аналитической концентрации 1.0 ммоль/л. Для упрощения заряды не включены в формулы анионов кислот.
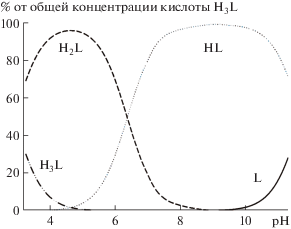
Константы устойчивости комплексов Cu2+ с депротонированными формами кислоты H3L определены методом потенциометрии с помощью программы CHEMEQUI (табл. 4). Согласно диаграмме распределения комплексов Cu2+ с кислотой H3L (рис. 3), в растворе образуются комплексы состава Cu : L = 1 : 2 и в гораздо меньшем количестве комплексы Cu : L = 1 : 1, что согласуется с преимущественной кристаллизацией комплекса [Cu(Н2L)2(Н2О)2]. При начальных концентрациях реагентов ~0.5 ммоль/л максимумы концентраций комплексов наблюдаются при pH 6.6 (CuL–) и pH 8.5 (${\text{CuL}}_{2}^{{4 - }}$). При pH > 8 происходит образование и рост доли гидроксилсодержащих комплексов Cu(OH)L2– и Cu(OH)${\text{L}}_{2}^{{5 - }}$ (рис. 3).
Таблица 4.
Ступенчатые и полные константы устойчивости комплексов Cu2+ с кислотой H3L в воде при температуре 298 K и ионной силе 0.1 M KCl
| i | Равновесие | lgKi ± sd | Равновесие | lgβi ± sd |
|---|---|---|---|---|
| 1 | Cu + L = CuL | 8.91 ± 0.06 | Cu + L = CuL | 8.91 ± 0.06 |
| 2 | CuL + L = CuL2 | 8.39 ± 0.08 | Cu + 2L = CuL2 | 17.30 ± 0.05 |
| 3 | CuL + OH = Cu(OH)L | 4.48 ± 0.12 | Cu + L + OH = Cu(OH)L | 13.39 ± 0.11 |
| 4 | CuL2 + OH = Cu(OH)L2 | 4.46 ± 0.44 | Cu + 2L + OH = Cu(OH)L2 | 21.76 ± 0.44 |
Рис. 3.
Диаграмма распределения комплексов Cu2+ с кислотой H3L в зависимости от pH в воде при 298 K, ионной силе 0.1 М и начальных концентрациях реагентов 0.69 (H3L) и 0.34 ммоль/л (Cu2+). Для упрощения заряды не включены в формулы, α – доля в процентах относительно общей концентрации Cu2+.
Первая из констант устойчивости (lgK1 = 8.91 и lgK2 = 8.39) комплексов CuL– и ${\text{CuL}}_{2}^{{4 - }}$ оказалась ниже соответствующей константы комплексов салициловой кислоты с Cu2+ (lgK1 = 10.83 и lgK2 = = 8.05 [52, 53]. Очевидно, это связано с тем, что фенольный кислород лиганда H3L в меньшей степени участвует в комплексообразовании, чем кислород фосфорильной группы. Согласно структурным данным, длины связей Cu–OPh (2.448(3) Å) и Cu–OP(O)(OH)Ph (1.967(3) Å) существенно различаются (см. рис. 1), т.е. эффект кооперативного связывания двумя координационными центрами лиганда незначительный, в отличие, например, от 4-метоксисалициловой кислоты, для которой связи Cu–OPh (1.899 Å) и Cu–OС(O)Ph (1.889 Å) близки по длине [54]. Тем не менее значение lgK1 комплекса Cu2+ с H3L (табл. 4) несколько выше соответствующих констант (lgKCuL = 8.51 [55], 7.69 [56], 7.27 [56]) комплексов Cu2+ с подандами 1,5-бис[2-(диоксифосфорил)-4-этилфенокси]-3-оксапентаном, 1,5-бис[2-(диоксифосфорил)фенокси]-3-оксапентаном и 1,8-бис[2-(диоксифосфорил)фенокси]-3,6-диоксаоктаном, являющимися алкилированными аналогами 2-оксифенилфосфоновой кислоты. Это косвенно указывает на определенное участие фенольной группы кислоты H3L в комплексообразовании с Cu2+ с учетом того, что, согласно данным РСА [44, 46], в связывании с катионом меди(II) участвуют только фосфорильные группы указанных подандов.
Предсказание устойчивости комплексов Cu2+ на основе ранее полученных моделей структура лиганда–устойчивость комплекса [57] дает согласующиеся с экспериментальными оценки констант lgK1 и lgK2 с учетом их погрешностей: 6.5 ± ± 1.1 (73) и 7.7 ± 1.1 (55). В скобках указано число моделей структура–свойство, вовлеченных в предсказание.
Известно, что одними из самых распространенных противовоспалительных препаратов являются производные салициловой кислоты, обладающие как противовоспалительным, так и анальгетическим действием [58]. Однако их передозировка может привести к серьезным побочным эффектам [59, 60]. Салицилатные комплексы Cu(II) оказались более эффективными лекарственными средствами с меньшим токсическим воздействием [61, 62]. Ранее [23] нами было показано, что 2-оксифенилфосфонистая кислота (L1) обладает низкой токсичностью и заметной анальгетической и противовоспалительной активностью. Поскольку биологическая активность H3L и [Cu(H2L)2(H2O)2] не исследовалась, была проведена оценка острой токсичности и анальгетической активности этих соединений.
Установлено, что при внутрижелудочном введении H3L и комплекса [Cu(H2L)2(H2O)2] для мышей обоего пола значения ЛД50 ≥ 2000 мг/кг. При внутрибрюшинном способе введения H3L существует гендерное различие, а именно: для мышей самок LD50 составляет 350 мг/кг, для самцов – 400 мг/кг. Тем не менее при обоих способах введения по своим основным токсикометрическим параметрам H3L относится к 4 классу токсичности – к малотоксичным соединениям.
Проверка анальгетической активности показала, что введение [Cu(H2L)2(H2O)2] вызывает у мышей дозозависимое уменьшение количества корчей, вызванных внутрижелудочным введением уксусной кислоты. Результаты проверки острой токсичности и анальгетической активности комплекса [Cu(H2L)2(H2O)2] в сравнении с H3L [11] и анальгином приведены в табл. 5.
Таблица 5.
Острая токсичность и анальгетическая активность Н3L, [Cu(H2L)2(H2O)2] и анальгина
| Соединение | LD50, мг/кг | Анальгетическая активность | |
|---|---|---|---|
| ED50, мг/кг | TI | ||
| H3L | 2000 | 70 | 28.6 |
| [Cu(H2L)2(H2O)2] | 2000 | 5.8 | 344.8 |
| Анальгин | 3390 | 42 | 80.7 |
Анализ результатов показал, что при малой токсичности анальгетический эффект комплекса [Cu(H2L)2(H2O)2] на моделях уксусных корчей значительно выше, чем у анальгина и свободной H3L, и даже выше анальгетического эффекта 2-оксифенилфосфонистой кислоты (L1) (ED50 = = 18; TI = 194), о которой говорилось в [11]. Результаты вскрытия лабораторных животных не показали ульцерогенного воздействия кислоты H3L в дозах, соответствующих ЕД50.
ЗАКЛЮЧЕНИЕ
Настоящая работа продолжает изучение 2-оксифенилфосфоновых кислот − фосфорильных аналогов салициловой кислоты. При комплексообразовании 2-окси-5-этилфенилфосфоновая кислота (H3L) выступает в роли сильной кислоты в качестве хелатирующего лиганда. В координации с катионом меди(ΙΙ) участвуют атомы кислорода фенольной и депротонированной фосфорильной групп. Впервые получены константы протонирования 2-окси-5-этилфосфоновой кислоты и константы устойчивости ее комплексов с Cu2+ в воде. Результаты биологических исследований показали, что комплекс [Cu(H2L)2(H2O)2] обладает высокой анальгетической активностью, при этом оба соединения не обладают ульцерогенным действием в дозах, соответствующих ЕД50. По своим основным токсикометрическим параметрам кислота H3L и комплекс [Cu(H2L)2(H2O)2] относятся к малотоксичным веществам.
Список литературы
Quin L.D. A Guide to Organophosphorus Chemistry. New York: Wiley-Interscience, 2000. 408 p.
Best Synthetic Methods: Organophosphorus (V) Chemistry / Ed. Timperley C.M. London: Academic Press, 2013. 786 p. https://doi.org/10.1016/C2011-0-04165-0
De Clercq E. // Biochem. Pharmacol. 2011. V. 82. № 2. P. 99. https://doi.org/10.1016/j.bcp.2011.03.027
Pradere U., Garnier-Amblard E.C., Coats S.J. et al. // Chem. Rev. 2014. V. 114. № 18. P. 9154. https://doi.org/10.1021/cr5002035
Queffélec C., Petit M., Janvier P. et al. // Chem. Rev. 2012. V. 112. № 7. P. 3777. https://doi.org/10.1021/cr2004212
Shameem M.A., Orthaber A. // Chem. - A Eur. J. 2016. V. 22. № 31. P. 10718. https://doi.org/10.1002/chem.201600005
Dutartre M., Bayardon J., Jugé S. // Chem. Soc. Rev. 2016. V. 45. № 20. P. 5771. https://doi.org/10.1039/C6CS00031B
Ivanova I.S., Ilyukhin A.B., Tsebrikova G.S. et al. // Inorg. Chim. Acta. 2019. V. 497. P. 119095.https://doi.org/10.1016/j.ica.2019.119095
Иванова И.С., Баулин В.E., Полякова И.Н. и др. // Журн. общ. химии. 2017. Т. 87. № 11. С. 1833. [Ivanova I.S., Baulin V.E., Polyakova I.N. et al. // Russ. J. Gen. Chem. 2017. V. 87. № 11. P. 2574. https://doi.org/10.1134/S107036321711010X]
Zhang M., Jia X., Zhu H. et al. // Org. Biomol. Chem. 2019. V. 17. № 11. P. 2972. https://doi.org/10.1039/C9OB00129H
Баулин В.Е., Калашникова И.П., Вихарев Ю.Б. и др. // Журн. общ. химии. 2018. Т. 88. № 9. С. 1438. [Baulin V.E., Kalashnikova I.P., Vikharev Y.B. et al. // Russ. J. Gen. Chem. 2018. V. 88. № 9. P. 1786. https://doi.org/10.1134/S1070363218090049].https://doi.org/10.1134/s0044460x18090044
Иванова И.С., Баулин В.Е., Пятова Е.Н. и др. // Журн. общ. химии. 2018. Т. 88. № 9. С. 1524. [Ivanova I.S., Baulin V.E., Pyatova E.N. et al. // Russ. J. Gen. Chem. 2018. V. 88. № 9. P. 1867. https://doi.org/10.1134/S1070363218090177]https://doi.org/10.1134/s0044460x18090172
Weder J.E., Dillon C.T., Hambley T.W. et al. // Coord. Chem. Rev. 2002. V. 232. № 1–2. P. 95. https://doi.org/10.1016/S0010-8545(02)00086-3
Novak E., Osborne D.W., Matheson L.E. et al. // Drug Dev. Ind. Pharm. 1991. V. 17. № 3. P. 373. https://doi.org/10.3109/03639049109043833
Hambley T.W. // Dalton Trans. 2007. № 43. P. 4929. https://doi.org/10.1039/b706075k
van Rijt S.H., Sadler P.J. // Drug Discov. Today. 2009. V. 14. № 23–24. P. 1089. https://doi.org/10.1016/j.drudis.2009.09.003
Ronconi L., Sadler P.J. // Coord. Chem. Rev. 2007. V. 251. № 13-14 spec. iss. P. 1633. https://doi.org/10.1016/j.ccr.2006.11.017
Thompson K.H., Orvig C. // Science. 2003. V. 300. № 5621. P. 936. https://doi.org/10.1126/science.1083004
Wehbe M., Leung A.W.Y., Abrams M.J. et al. // Dalton Trans. 2017. V. 46. № 33. P. 10758. https://doi.org/10.1039/c7dt01955f
Eshaghi Malekshah R., Salehi M., Kubicki M. et al. // J. Coord. Chem. 2018. V. 71. № 7. P. 952. https://doi.org/10.1080/00958972.2018.1447668
Sadhu M.H., Kumar S.B., Saini J.K. et al. // Inorg. Chim. Acta. 2017. V. 466. P. 219. https://doi.org/10.1016/j.ica.2017.06.006
Ndagi U., Mhlongo N., Soliman M.E. // Drug Des. Devel. Ther. 2017. V. 11. P. 599. https://doi.org/10.2147/DDDT.S119488
Shabbir M., Akhter Z., Ismail H. et al. // J. Mol. Struct. 2017. V. 1146. P. 57. https://doi.org/10.1016/j.molstruc.2017.05.127
Piri Z., Moradi-Shoeili Z., Assoud A. // Inorg. Chem. Commun. 2017. V. 84. P. 122. https://doi.org/10.1016/j.inoche.2017.08.005
Jayamani A., Sengottuvelan N., Chakkaravarthi G. // Polyhedron. 2014. V. 81. P. 764. https://doi.org/10.1016/j.poly.2014.05.076
Uzun N., Colak A.T., Emen F.M. et al. // J. Coord. Chem. 2015. V. 68. № 6. P. 949. https://doi.org/10.1080/00958972.2014.1003371
Ling X., Cutler C.S., Anderson C.J. The Radiopharmaceutical Chemistry of the Radioisotopes of Copper Springer Nature Switzerland AG 2019, 2019. P. 335. https://doi.org/10.1007/978-3-319-98947-1_19
Bandara N., Sharma A.K., Krieger S. et al. // J. Am. Chem. Soc. 2017. V. 139. № 36. P. 12550. https://doi.org/10.1021/jacs.7b05937
Орлова М.А., Трофимова Т.П., Золотова Н.С. и др. // Изв. АН Сер. хим. 2019. № 10. С. 1933. [Orlova M.A., Trofimova T.P., Zolotova N.S. et al. // Russ. Chem. Bull. Int. Ed. 2019. V. 68. № 10. P. 1933. https://doi.org/10.1007/s11172-019-2649-2]
McInnes L.E., Noor A., Kysenius K. et al. // Inorg. Chem. 2019. V. 58. № 5. https://doi.org/10.1021/acs.inorgchem.8b03466
Shuvaev S., Kotova O., Utochnikova V. et al. // Inorg. Chem. Commun. 2012. V. 20. P. 73. https://doi.org/10.1016/j.inoche.2012.02.020
Shuvaev S., Bushmarinov I.S., Sinev I. et al. // Eur. J. Inorg. Chem. 2013. № 27. P. 4823. https://doi.org/10.1002/ejic.201300540
Цебрикова Г.С., Барсамян Р.Т., Соловьев В.П. и др. // Изв. АH Сер. хим. 2018. № 12. С. 2184. [Tsebrikova G.S., Barsamian R.T., Solov’ev V.P. et al. // Russ. Chem. Bull. Int. Ed. 2018. V. 67. № 12. P. 2184. https://doi.org/10.1007/s11172-018-2352-8]
Соловьев В.П. Программа ChemEqui для расчета констант химических равновесий и сопутствующих параметров, исходя из экспериментальных результатов физико-химических методов, таких как УФ, ИК и ЯМР спектроскопия, калориметрия, потенциометрия и кондуктометрия. http://vpsolovev.ru/programs/ (12 августа 2020).
Solov’ev V.P., Tsivadze A.Y. // Prot. Met. Phys. Chem. Surfaces. 2015. V. 51. № 1. P. 1. https://doi.org/10.1134/S2070205115010153
Muller P.H., Neumann P., Storm R. Tafeln der mathematischen Statistik. Leipzip: VEB Fachbuchverlag, 1979.
Bandyopadhyay S., Das A., Mukherjee G.N. et al. // Inorg. Chim. Acta. 2004. V. 357. № 12. P. 3563. https://doi.org/10.1016/j.ica.2004.05.010
Ali M., Pant M., Abraham A. // Trans. Inst. Meas. Control. 2012. V. 34. № 6. P. 691. https://doi.org/10.1177/0142331211403032
Bruker AXS Inc. // APEX3 and SAINT 2016.
Sheldrick G.M. // SADABS, Programs Scaling Absorpt. Correct. Area Detect. Data. 1997.
Sheldrick G.M. // Acta Crystallogr., Sect. C: Struct. Chem. 2015. V. 71. № 1. P. 3. https://doi.org/10.1107/S2053229614024218
Беленький М.Л. Элементы количественной оценки фармакологического эффекта. Л.: Медицинская литература, 1963. 146 с.
Tsebrikova G.S., Polyakova I.N., Solov’ev V.P. et al. // Inorg. Chim. Acta. 2018. V. 478. P. 250. https://doi.org/10.1016/j.ica.2018.04.007
Баулин В.E., Кискин М.А., Иванова И.С. и др. // Журн. неорган. химии. 2012. Т. 57. № 5. С. 739. [Baulin B.E., Kiskin M.A., Ivanova I.S. et al. // Russ. J. Inorg. Chem. 2012. V. 57. № 5. P. 671. https://doi.org/10.1134/S0036023612050038]
Баулин В.E., Миначева Л.Х., Иванова И.С. и др. // Журн. неорган. химии. 2011. Т. 56. № 8. С. 1293. [Baulin V.E., Minacheva L.K., Ivanova I.S. et al. // Russ. J. Inorg. Chem. 2011. V. 56. № 8. P. 1222. https://doi.org/10.1134/S0036023611080043]
Баулин В.E., Миначева Л.Х., Иванова И.С. и др. // Журн. неорган. химии. 2011. Т. 56. № 8. С. 1303. [Baulin V.E., Minacheva L.K., Ivanova I.S. et al. // Russ. J. Inorg. Chem. 2011. V. 56. № 8. P. 1232. https://doi.org/10.1134/S0036023611080055]
Демин С.В., Нефедов С.Е., Баулин В.Е. и др. // Коорд. химия. 2013. Т. 39. № 4. С. 223. [Demin S.V., Nefedov S.E., Baulin V.E. et al. // Russ. J. Coord. Chem. 2013. V. 39. № 4. P. 333. https://doi.org/10.1134/S1070328413040052]https://doi.org/10.7868/s0132344x13040051
Полякова И.Н., Баулин В.Е., Иванова И.С. и др. // Кристаллография. 2015. Т. 60. № 1. С. 63. [Polyakova I.N., Baulin V.E., Ivanova I.S. et al. // Crystallogr. Reports. 2015. V. 60. № 1. P. 57. https://doi.org/10.1134/S1063774515010162]https://doi.org/10.7868/s0023476115010166
Беллами Л. Инфракрасные спектры сложных молекул. М.: Изд-во иностр. литер., 1963. 590 с. [Bellamy L.J. The Infra-Red Spectra of Complex Molecules. London–New York: Methuen & Co. LTD, JHN Wiley & Sons, Inc., 1954.]
Наканиси К. Инфракрасные спектры и строение органических соединений. М.: Мир, 1965. 216 c. [Nakanishi K. Infrared Absorption Spectroscopy. San Francisco: Holden-Day, Inc.; Tokyo: Nankodo Company Limited, 1962].
Nualláin C.Ó. // J. Inorg. Nucl. Chem. 1974. V. 36. № 2. P. 339. https://doi.org/10.1016/0022-1902(74)80020-5
Lajunen L.H.J., Portanova R., Piispanen J. et al. // Pure Appl. Chem. 1997. V. 69. № 2. P. 329. https://doi.org/10.1351/pac199769020329
Venkatnarayana G., Swamy S., Lingaiah P. // Indian J. Chem. 1984. V. 23A. № 6. P. 501.
Puchoňová M., Matejová S., Jorík V. et al. // Polyhedron. 2018. V. 151. P. 152. https://doi.org/10.1016/j.poly.2018.05.036
Аль Ансари Я.Ф., Баулин В.Е. // Журн. неорган. химии. 2019. Т. 64. № 4. С. 445. [Al Ansari Y.F., Baulin V.E. // Russ. J. Inorg. Chem. 2019. V. 64. № 4. P. 550. https://doi.org/10.1134/S0036023619040028]https://doi.org/10.1134/s0044457x19040020
Игнатьева Т.И., Баулин В.Е., Цветков Е.Н., Раевский О.А. // Журн. общ. химии. 1990. Т. 60. № 7. С. 1503.
Solov’ev V., Varnek A., Tsivadze A. // J. Comput. Aided. Mol. Des. 2014. V. 28. № 5. P. 549. https://doi.org/10.1007/s10822-014-9741-3
Doutremepuich C. // Thrombosis. 2012. V. 2012. Special Issue. P. 1. https://doi.org/10.1155/2012/626289
Turnbull C.M., Rossi A.G., Megson I.L. // Expert Opin. Ther. Targets. 2006. V. 10. № 6. P. 911. https://doi.org/10.1517/14728222.10.6.911
Buttgereit F., Burmester G.R., Simon L.S. // Am. J. Med. 2001. V. 110. № 3 Suppl. 1. P. 13. https://doi.org/10.1016/s0002-9343(00)00728-2
Jacka T., Bernard C.C.A., Singer G. // Life Sci. 1983. V. 32. № 9. P. 1023. https://doi.org/10.1016/0024-3205(83)90934-7
O’Connor M., Kellett A., McCann M. et al. // J. Med. Chem. 2012. V. 55. № 5. P. 1957. https://doi.org/10.1021/jm201041d
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии