

Журнал неорганической химии, 2021, T. 66, № 1, стр. 69-80
Предсказательное моделирование молекул высокоэнергетических гетероциклических веществ
В. М. Волохов a, *, Т. С. Зюбина a, А. В. Волохов a, Е. С. Амосова a, Д. А. Варламов a, Д. Б. Лемперт a, Л. С. Яновский a, b, c
a Институт проблем химической физики РАН
142432 Московская обл., Черноголовка, пр-т Академика Семенова, 1, Россия
b Центральный институт авиационного моторостроения им. П.И. Баранова
111116 Москва, ул. Авиамоторная, 2, Россия
c Московский авиационный институт (национальный исследовательский университет)
125993 Москва, Волоколамское шоссе, 4, Россия
* E-mail: vvm@icp.ac.ru
Поступила в редакцию 26.05.2020
После доработки 03.08.2020
Принята к публикации 27.08.2020
Аннотация
Оценка термохимических свойств ряда газообразных веществ (кДж/кг): C4N8O6 (2854.0), C4HN7O4 (2932.3), C4H2N6O2 (3104.8), C2N6O4 (4252.6), C2N8O4 (4395.9), C2N6O3 (4645.6), C2N8O4 (4755.2), C2HN7O2 (4815.7), C4N6O2 (5153.2), C4N8O2 (5609.1) проведена в рамках программного комплекса GAUSSIAN-09 с использованием метода DFT с гибридным функционалом плотности B3LYP на уровне B3LYP/6-311+G(2d,p) и комбинированных методов G4(MP2) и G4. Для экспериментально изученных веществ C2N6O4, C2N6O3, C2N8O4 рассчитанные на уровне G4 значения энтальпии ${{\Delta }_{f}}H_{{298}}^{^\circ }\left( {\text{г}} \right)$ больше экспериментальных на 8–15%, что почти вдвое меньше разброса экспериментальных значений для этих соединений. Термохимические данные, ИК-спектры поглощения, структурные параметры и смещения атомов для наиболее интенсивных колебаний высокоэнергетических соединений C4N8O6, C4HN7O4, C4H2N6O2, C2N8O4, C2HN7O2, C4N6O2, C4N8O2 получены впервые. Продемонстрировано, что использование уровня расчета G4(MP2) для выбранного класса молекул дает очень близкие (в пределах 1–3%) относительно уровня G4 результаты и ведет к существенной (в 3–8 раз) экономии расчетного времени. Однако результаты на уровне B3LYP/6-311+G(2d,p) отличаются от G4 на 2–11%. Полученные данные могут быть использованы как справочные и позволяют выделить наиболее перспективные группы веществ для работы в качестве высокоэнергетических компонент перспективного топлива.
ВВЕДЕНИЕ
Большой интерес к созданию высокоэнергетических материалов различного назначения стимулируется бурно развивающимися новыми технологиями, особенно в области двигателей перспективных летательных аппаратов. В последнее время в создании новых материалов с определенными свойствами все более важную роль играют современные компьютерные технологии [1–3].
Поскольку одним из основных критериев энергоемкости химического соединения является его стандартная энтальпия образования ΔfH°, определение этой величины, как экспериментальное, так и расчетное, становится ключевой задачей для оценки эффективности применения того или иного вещества как компонента высокоэнергетических материалов, таких как взрывчатые вещества, ракетное топливо, порох, пиротехнические композиции и пр. В задачах термохимии важно знать, как меняется энтальпия образования (ЭО) вещества при небольших изменениях структуры. Как правило, замещение определенного фрагмента в молекуле на другой сопровождается изменением ЭО. Но часто окружение атома сильно влияет на величины ЭО, поэтому применять широко известные так называемые аддитивные величины ЭО различных фрагментов, чтобы оценить ЭО конкретного соединения, в ряде случаев ненадежно. Желательно знать заранее закономерности изменения ЭО при замещении одного молекулярного фрагмента другим в рядах структурно близких по строению соединений.
Энтальпия образования ΔfH° в том агрегатном состоянии, в котором исследуемое вещество намечено к применению является основным параметром, характеризующим энергоемкость вещества. Эксплуатационные характеристики (удельный импульс топлива, дальность полета и другие) энергоемких соединений и композиций на их основе в достаточно широком диапазоне значений приблизительно линейно зависят от изменения ΔfH°. Для получения наиболее достоверной оценки перспективности того или иного соединения в энергетических составах различного назначения необходимо иметь как можно более точные величины ΔfH° компонентов, особенно присутствующих в композиции в качестве основного вещества. Из экспериментальных методов определения ЭО на сегодня практически единственным является сжигание вещества в калориметре: исследователи определяют удельную теплоту сгорания в кислороде, после чего рассчитывают ЭО. Калориметрическое измерение собственно теплового эффекта на современном оборудовании осуществляется с высокой точностью при условии 100%-го сгорания вещества до воды, азота и CO2. Часто наряду с азотом в продуктах сгорания образуются и оксиды азота, что требует их количественного определения и ввода поправки на теплоту их образования. Самые прецизионные эксперименты обычно дают средние величины ЭО с точностью 40–50 кДж/кг. При экспериментальном определении ЭО большой проблемой является недостаточно высокая чистота вещества. При этом, как правило, заранее неизвестно, какие микропримеси присутствуют в образце и каково их количество. Например, наличие в качестве примеси 0.1% воды при калориметрическом измерении ЭО октогена (ΔfH° = 295.15 кДж/кг) дает ошибку 9.5 кДж/кг, что составляет 3% от ЭО октогена, а если загрязнением является вещество с большей удельной теплотой сгорания, чем октоген, например бензол, то каждый 0.1% примеси бензола повысит ЭО на 32.3 кДж/кг, что составляет 11%.
Приведенные выше примеры свидетельствуют о важности применения прямых методов расчета энергетических и термохимических параметров высокоэнергетических веществ, таких как квантово-химические расчеты на основе ab initio подходов. Кроме того, квантово-химические методы позволяют осуществлять компьютерный дизайн и с высокой точностью рассчитывать предполагаемые термохимические параметры гипотетических, еще не синтезированных соединений. Это позволяет существенно сузить круг веществ, намеченных к синтезу, и ускорить процесс создания новых видов топлива.
В работе [4] на основании расчетов 454 газообразных веществ проведено подробное сравнение точности метода G4 с методами G2 и G3, а также с экспериментальными данными. Показано, что усредненное отклонение результатов расчетов методом G4 от экспериментальных данных составляет 0.83 ккал/моль. В работе [5] комбинированным методом W4-17 рассчитаны реакции атомизации 200 молекул и радикалов, состоящих из атомов первого и второго ряда, в состав которых входит не более восьми неводородных атомов. Анализ рассмотренных соединений показал, что комбинированные методы G4, Wn, Wn-F12, WnX, ccCA-PS3 обеспечивают среднеквадратичное отклонение расчета от эксперимента от 2.3 до 4.0 кДж/моль, в то время как для методов G3, Gn(MP2) и CBS оно превышает 4.2 кДж/моль. Комбинированный метод G4 дает наилучшие результаты (среднеквадратичные отклонения (СКО) равны 4.0 кДж/моль), однако для высокофторированных и хлорированных систем, таких как SF6, PF5, PF3, BF3, CCl4, C2Cl2, C2Cl4 и C2Cl6, эти отклонения могут достигать 20 кДж/моль. Метод G4(MP2) является наилучшим из комбинированных методов типа Gn(MP2), СКО равны 5.4 кДж/моль. Методы G4(MP2)-6X, ROG4(MP2)-6X, G3(MP2) и G3(MP2)B3 не так хороши, для них СКО находятся в интервале от 6.9 до 9.3 кДж/моль. Комбинированные методы типа CBS (CBS-QB3, ROCBS-QB3 и CBS-4M) приводят к среднеквадратичным отклонениям в диапазоне 8.5–17.7 кДж/моль. Метод ccCA-PS3 дает результаты, близкие к полученным методом G4 (СКО равны 3.8 кДж/моль). Использование метода W1 приводит к снижению СКО до 3.1 кДж/моль. Методы W1-F12 и W1X-n дают сопоставимые с W1 результаты при значительно уменьшенном вычислительном времени – они приводят к СКО между 2.8 (W1X-1) и 3.3 (W1X-2) кДж/моль. Более дорогие вычислительные методы W2-типа дают несколько лучшие результаты с СКО 2.3 (W2-F12), 2.4 (W2) и 2.6 (W2X) кДж/моль. Однако методы типа Wn пока доступны лишь для малых молекул, поэтому в качестве рабочих мы выбрали комбинированные методы G4 и G4(MP2).
Для исследования энергетических возможностей высокоэнтальпийных полиазотистых соединений как компонентов твердого ракетного топлива необходимо знать ΔfH° соединения в том фазовом состоянии, в каком оно должно находиться в композиции (чаще всего в твердой фазе). Для перевода параметров вещества из газообразного в твердое состояние необходимо знать энергию сублимации (ΔsubH°). Классический метод измерения ΔsubH° (или энтальпии парообразования) – это измерение температурной зависимости давления насыщенных паров испытуемого образца (ln p = A + exp(ΔfH°/(RT)), но пригодное для точного измерения давление паров многих энергоемких соединений возникает уже при температурах, когда начинается заметное разложение. Кроме того, этот метод неприменим, пока вещество не синтезировано реально, поэтому непригоден для предсказания и прогнозов. Однако можно воспользоваться различными методами оценки энергии сублимации, например, предложенными в работах [6] и [7]. Это позволяет переходить при расчете энтальпии образования вещества от газообразного к кристаллическому состоянию (и обратно).
Для расчета энтальпии образования ΔfH° газообразных молекул существует несколько методов различной сложности и точности. Среди них эмпирические и полуэмпирические методы, опирающиеся на предположение об аддитивности энтальпии образования сложной молекулы из групп атомов, энтальпии образования которых известны [8]. Используются также методы, опирающиеся на DFT-расчеты [9–15]. Расчеты на базе комбинированного метода G3 [16–18], проведенные в работах [19–30] при исследовании распада структур типа CmHn, демонстрируют точность воспроизведения сравнительных энергий в пределах 10 кДж/моль.
В настоящей работе проведен расчет структуры, энтальпии образования и ИК-спектров десяти газообразных молекул ряда высокоэнтальпийных производных гетероциклов на базе фуразана, фуроксана и 1,2,3-триазола: (1) C4N8O6 (4,8-динитро-4H,8H-бис([1, 2, 5]оксадиазоло)[3,4-b:3',4'-e]пиразин), (2) C4HN7O4 (4-нитро-4H,8H-бис([1, 2, 5]оксадиазоло)[3,4-b:3',4'-e]пиразин), (3) C4H2N6O2 (4H,8H-бис([1, 2, 5]оксадиазоло)[3,4-b:3',4'-e]пиразин), (4) C2N6O4 ([1, 2, 5]оксадиазоло[3,4-е] [1–4]тетразин-1,4,6-триоксид), (5) C2N8O4 (1-нитро-1H- [1–3]триазоло[4,5-е] [1–4]тетразин-5,7-диоксид), (6) C2N6O3 ([1, 2, 5]оксадиазоло[3,4-е] [1–4]тетразин-4,6-диоксид), (7) C2N8O4 ([1–4]тетразино[5,6-e] [1–4]тетразин-1,3,6,8-тетраоксид), (8) C2HN7O2 (1H- [1–3]триазоло[4,5-е] [1–4]тетразин-5,7-диоксид), (9) триплет C4N6O2, (10) C4N8O2 ((4Z,9Z)-бис([1, 2, 5]оксадиазоло)[3,4-c:3',4'-g] [1, 2, 5, 6]тетразоцин).
В табл. 1 приведены полученные из литературных источников энергии сублимации и энтальпии трех известных веществ: C2N6O4, C2N6O3 и C2N8O4 в газообразном и кристаллическом состоянии. Видно, что разница в экспериментально полученных значениях ${{\Delta }_{f}}H_{{298}}^{^\circ }\left( {\text{г}} \right)$ достаточно велика (от 10 до 30% от наибольшего значения). В связи с этим возникает вопрос о дальнейшем уточнении этих результатов (например, квантово-химическими методами). Поэтому кроме трех известных, приведенных в табл. 1, нами рассчитаны семь новых, еще не синтезированных веществ (табл. 2), структуры которых приведены на рис. 1.
Таблица 1.
Полученные из литературных источников энергии сублимации (${{\Delta }_{{{\text{sub}}}}}H_{{298}}^{^\circ }$, кДж/кг) и энтальпии C2N6O4(структура 4, табл. 2), C2N6O3(структура 6, табл. 2) и C2N8O4(структура 7, табл. 2) в газообразном (${{\Delta }_{f}}H_{{298}}^{^\circ }$(г), кДж/кг) и кристаллическом состоянии (${{\Delta }_{f}}H_{{298}}^{^\circ }$(кр), кДж/кг)
| № | Формула | ${{\Delta }_{f}}H_{{298}}^{^\circ }$(г), кДж/кг | ${{\Delta }_{{{\text{sub}}}}}H_{{298}}^{^\circ }$, кДж/кг | ${{\Delta }_{f}}H_{{298}}^{^\circ }$(кр), кДж/кг |
|---|---|---|---|---|
| 4 | C2N6O4 | 3051.818 ± 16.72 [1] 3678.4 [3] |
331.892 ± 83.6 [1] | 2719.926 ± 83.6 [1] 2675.2 [3] |
| 6 | C2N6O3 | 3031.336 ± 20.9 [1] 4321 [4] 4311 [5] |
268.356 ± 83.6 [1] 264.176 ± 14.212 [2] |
2762.98 ± 87.78 [1], 2813.976 ± 40.128 [5] |
| 7 | C2N8O4 | 3976.016 ± 25.08 [1] 4316 [6] [7] |
359.898 ± 83.6 [1] | 3616.118 ± 87.78 [1] |
Таблица 2.
Структурные формулы, молекулярные веса и энтальпии рассчитанных молекул, полученные на разных уровнях расчета*
| № | Формула М, а.е.м. | Энтальпия | Уровень расчета | ||
|---|---|---|---|---|---|
| ккал/моль | кДж/моль | кДж/кг | |||
| 1 | C4N8O6 256.094 |
199.7 | 835.9 | 3264.0 | B3LYP/6-311+G(2d,p) |
| 180.9 | 757.2 | 2956.7 | G4(MP2) | ||
| 174.6 | 730.9 | 2854.0 | G4 | ||
| 2 | C4HN7O4 211.097 |
166.8 | 698.0 | 3306.5 | B3LYP/6-311+G(2d,p) |
| 152.9 | 639.7 | 3030.4 | G4(MP2) | ||
| 147.9 | 619.0 | 2932.3 | G4 | ||
| 3 | C4H2N6O2 166.100 |
136.3 | 570.6 | 3435.3 | B3LYP/6-311+G(2d,p) |
| 126.8 | 530.8 | 3195.7 | G4(MP2) | ||
| 123.2 | 515.7 | 3104.8 | G4 | ||
| 4 | C2N6O4 172.060 |
180.7 | 756.4 | 4396.1 | B3LYP/6-311+G(2d,p) |
| 178.7 | 748.0 | 4347.3 | G4(MP2) | ||
| 174.8 | 731.7 | 4252.6 | G4 | ||
| 5 | C2N8O4 200.074 |
218.8 | 915.9 | 4577.8 | B3LYP/6-311+G(2d,p) |
| 214.5 | 897.9 | 4487.8 | G4(MP2) | ||
| 210.1 | 879.5 | 4395. 9 | G4 | ||
| 6 | C2N6O3 156.061 |
180.3 | 754.7 | 4835.9 | B3LYP/6-311+G(2d,p) |
| 176.3 | 738.0 | 4728.9 | G4(MP2) | ||
| 173.2 | 725.0 | 4645. 6 | G4 | ||
| 7 | C2N8O4 200.074 |
231.3 | 967.9 | 4837.7 | B3LYP/6-311+G(2d,p) |
| 231.8 | 970.0 | 4848.2 | G4(MP2) | ||
| 227.4 | 951.4 | 4755.2 | G4 | ||
| 8 | C2HN7O2 155.077 |
182.7 | 764.8 | 4931.7 | B3LYP/6-311+G(2d,p) |
| 181.2 | 758.5 | 4891.1 | G4(MP2) | ||
| 178.4 | 746.8 | 4815.7 | G4 | ||
| 9 | C4N6O2 164.084 |
206.6 | 864.3 | 5267.4 | B3LYP/6-311+G(2d,p) |
| 204.0 | 853.7 | 5203.0 | G4(MP2) | ||
| 202.1 | 845.6 | 5153.2 | G4 | ||
| 10 | C4N8O2 192.098 |
272.8 | 1141.9 | 5944.4 | B3LYP/6-311+G(2d,p) |
| 260.1 | 1088.8 | 5667.9 | G4(MP2) | ||
| 257.4 | 1077.5 | 5609.1 | G4 | ||
* Для расчета использованы экспериментальные атомные энтальпии образования из NIST-JANAFThermochemicaltables [33]: ${{\Delta }_{f}}H_{{298}}^{^\circ }$ = 171.288 ± 0.108 (C), ${{\Delta }_{f}}H_{{298}}^{^\circ }$ = 112.974 ± 0.096 (N), ${{\Delta }_{f}}H_{{298}}^{^\circ }$ = 59.554 ± 0.024 (O) и ${{\Delta }_{f}}H_{{298}}^{^\circ }$ = 52.103 ± 0.001 (H) (ккал/моль).
МЕТОДИКА РАСЧЕТА
В данной работе расчет энтальпии образования был проведен методом атомизации исследуемых молекул с применением комбинированных методов G4 и G4(MP2) [4, 31, 32]. Ниже на примере молекулы CmHn продемонстрированы основные принципы расчета энтальпии образования методом атомизации газообразных молекул. При этом выполняется наиболее естественная и удобная, на наш взгляд, последовательность расчетов.
На первом этапе рассчитывается энергия атомизации в нерелятивистском приближении: $\sum {{{D}_{0}}} = m{{E}_{e}}(C) + n{{E}_{e}}(H) - {{E}_{0}}({{C}_{m}}{{H}_{n}})$, где ${{E}_{e}}(C),{{E}_{e}}(H)$ – рассчитанные полные энергии атомов углерода и водорода. Полную энергию молекулы ${{E}_{0}}({{C}_{m}}{{H}_{n}})$ рассчитывают по формуле ${{E}_{0}}({{C}_{m}}{{H}_{n}}) = {{\varepsilon }_{0}} + {\text{ZPE}}$, где ɛ0 – полная энергия молекулы, а ZPE – сумма энергий нулевых колебаний всех колебательных мод молекулы. Энтальпия образования молекулы при 0 K рассчитывается по формуле $\Delta H_{f}^{0}({{C}_{m}}{{H}_{n}},0K)$ = $m\Delta H_{f}^{0}(C,0K)$ + + $n\Delta H_{f}^{0}(H,0K) - \sum {{{D}_{0}}} $. Первые два слагаемых – это энтальпии образования газообразных атомных компонент, известные из эксперимента. Энтальпия образования молекулы при 298.15 K вычисляется как $\Delta H_{f}^{0}({{C}_{m}}{{H}_{n}},298\,{\text{K}})$ = $\Delta H_{f}^{0}({{C}_{m}}{{H}_{n}},0\,{\text{K}})$ + + $\left( {{{H}^{0}}({{C}_{m}}{{H}_{n}},298\,{\text{K}}) - {{H}^{0}}({{C}_{m}}{{H}_{n}},0\,{\text{K}})} \right)$ – $m\left( {{{H}^{0}}(C,298\,{\text{K}})} \right.$ – $\left. {{{H}^{0}}(C,0\,{\text{K}})} \right)$ – $n\left( {{{H}^{0}}(H,298\,{\text{K}})} \right.$ – ‒ $\left. {{{H}^{0}}(H,0\,{\text{K}})} \right)$. Второе слагаемое получается из расчета молекулы. Третий и четвертый члены в последнем уравнении известны из эксперимента (или рассчитаны по экспериментальным молекулярным постоянным). Значения энтальпии образования газообразных атомов и термические поправки могут быть взяты из различных справочников или литературных источников, например [32–36]. В настоящей работе мы использовали экспериментальные атомные энтальпии образования из термохимических таблиц NIST-JANAF [33]. Поскольку теоретический расчет систематически завышает величины частот нулевых колебаний, существует практика коррекции частот с использованием эмпирически подобранных коэффициентов. Как и при рассмотрении самих частот, для получения более точных результатов необходимо корректировать частоты колебаний при расчете поправок ZPE и $\left( {{{H}^{0}}({{C}_{m}}{{H}_{n}},298\,{\text{K}}) - {{H}^{0}}({{C}_{m}}{{H}_{n}},0\,{\text{K}})} \right)$. Для этого используются значения масштабирующих множителей, рекомендуемые в литературе для различных методов расчетов и различных базисных наборов [37].
Однако описание энергий реакций требует использования теории высокого уровня, которая бы адекватно учитывала эффект электронной корреляции. В связи с этим особое внимание было уделено выбору наиболее эффективных методов квантово-химических расчетов, сочетающих точность расчетов и оптимальные временные затраты на вычисления. Специально для термохимических расчетов в последние десятилетия был разработан и усовершенствован ряд комбинированных методов, из которых наиболее известными являются методы семейства Gaussian-N (Gn) [31]. В них расчет высокого уровня аппроксимируется серией расчетов более низкого уровня с целью достижения более высокой точности в предсказании термохимических характеристик системы. В настоящей работе был использован метод G4 (Gaussian-4), представленный в 2007 г. Кертиссом и коллегами [4]. В этой работе показано, что метод G4 по сравнению с G3 [16, 38] значительно улучшает среднеквадратичное отклонение (от 1.13 ккал/моль для G3 до 0.83 ккал/моль для G4). Во многом это определило выбор данного метода для проведения наших квантово-химических расчетов.
В методе G4 для аппроксимации энергий более точных расчетов метод CCSD(Т) с достаточно высоким уровнем корреляции электронов и базисным набором среднего размера (6-31G(d)) сочетается с энергиями из расчетов более низкого уровня теории (MP4 и MP2) с большими базисными наборами. Кроме того, в оценку оставшихся погрешностей включены эмпирические поправки, не зависящие от изучаемой молекулы. Существует несколько модификаций метода G4, из которых в настоящей работе использован метод G4(МР2) [31]. В этом случае расчет МР4 заменяется на МР2. Метод G4(МР2) менее требователен к компьютерным ресурсам, поэтому он может использоваться в тех случаях, когда расчет G4 не может быть проведен из-за ограниченности вычислительных ресурсов. Этот метод имеет меньшую точность по сравнению с методом G4. Для тестирования метода его разработчики использовали тестовый набор, содержащий 270 экспериментальных энтальпий образования. Общее среднее абсолютное отклонение (САО) в этом тестовом наборе составляло 3.3 кДж/моль для G4 и 4.1 кДж/моль для G4(МР2) методов. Авторы [39] представили результаты расчетов ${{\Delta }_{f}}H_{{298}}^{^\circ }\left( {\text{г}} \right)$ для 63 азотсодержащих соединений (амины, амиды, нитроэфиры, нитросоединения и др.). Стандартное отклонение в случае метода G4 составляло 3.1 кДж/моль. Согласно [31], при расчете энтальпий для G4 САО составляет 3.1 кДж/моль, а для G4(MP2) – 3.9 кДж/моль. В настоящее время метод G4 позволяет получить самые надежные значения энтальпии образования методом реакций атомизации для крупных (более 10 неводородных атомов) органических молекул.
В нашей предыдущей работе [40] показано, что использование уровня расчета G4(MP2) для молекул типа CmHn (m = 10, 16, 18, 19, 26, n = 6, 8, 10) дает очень близкие (в пределах 1%) относительно уровня G4 результаты и ведет к существенной (в 3–8 раз) экономии расчетного времени. Для экспериментально измеренного соединения 1,4-диэтинилбензола (C10H6) было получено отклонение расчетного значения ЭО от экспериментального 2–3% на уровнях G4(MP2) и G4.
В настоящей работе моделирование было выполнено в рамках программного комплекса GAUSSIAN 09 [37] с использованием хорошо зарекомендовавшего себя в молекулярных расчетах гибридного функционала плотности B3LYP [41, 42] с базисом 6-311+G(2d,p) и комбинированных методов G4 и G4(MP2) [4, 31].
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
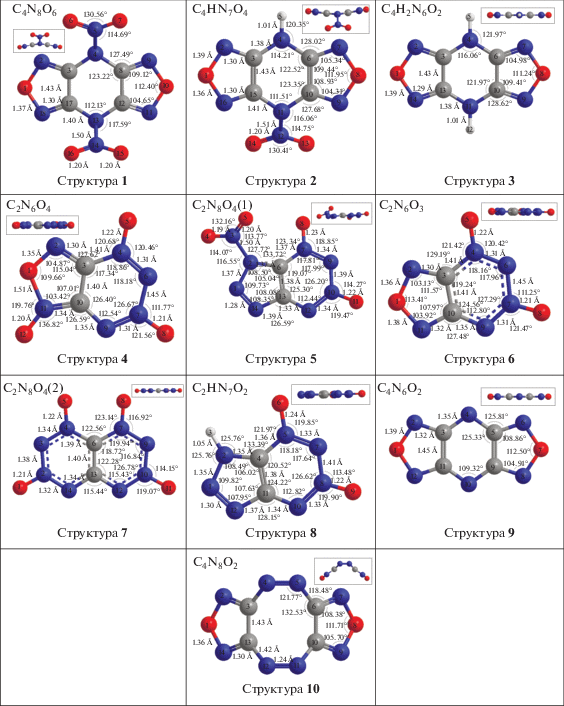
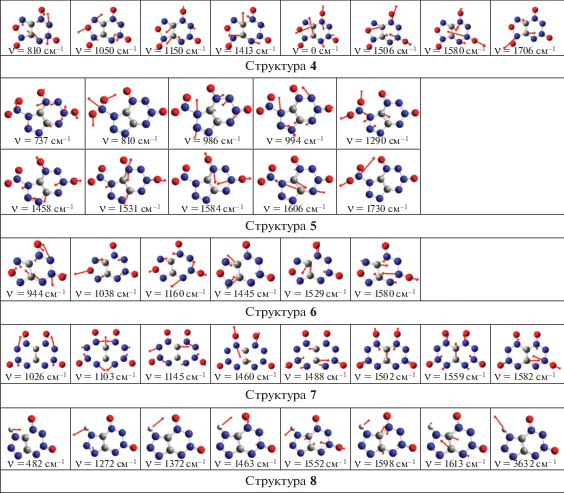
Энтальпии образования. Результаты расчетов показаны в табл. 2 и на рис. 1–7, где приведены структуры, геометрические параметры, ИК-спектры поглощения, смещения атомов и энтальпии образования рассчитанных газообразных молекул, полученные на разных уровнях расчета. В целом, приведенные в табл. 2 значения демонстрируют незначительные различия (в пределах 1–3%) в результатах, полученных на уровнях G4 и G4(MP2). Однако результаты, полученные на уровне B3LYP/6-311+G(2d,p), отличаются от наиболее точных, полученных на уровне G4, на 2–11%. При этом наибольшие отклонения наблюдаются для молекул типа (C2N2O–R1)2 (R1 = N2O2, NH, N и N2, структуры 1–3, 9 и 10, рис. 2), а наименьшие – для молекул типа C2N4O2–R2 (R2 = NONO, N4O2, NON, N2ON2O, N3H, структуры 4–8, рис. 3).
Рис. 2.
Относительные изменения энтальпий образования структур 1–3 и 9–10. Черные квадраты – уровень расчета B3LYP/6-311+G(2d,p), красные круги – уровень расчета G4(MP2), синие треугольники – уровень расчета G4.
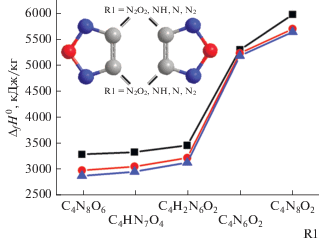
Рис. 3.
Относительные изменения энтальпий образования структур 4–8. Черные квадраты – уровень расчета B3LYP/6-311+G(2d,p), красные круги – уровень расчета G4(MP2), синие треугольники – уровень расчета G4.
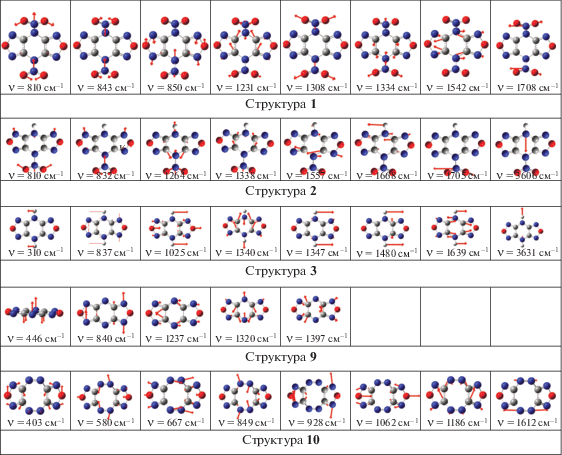
Из табл. 2 и рис. 2, 3 видно, что в случае сшивки фуразановых пятичленных колец C2N2O фрагментами R1 = N2O2, NH, N и N2 (структуры 1–3, 9 и 10 соответственно) энтальпия возрастает (2854.0, 2932.3, 3104.8, 5153.2 и 5609.1 кДж/кг соответственно) с ростом массовой доли азота во фрагменте R1 (от 23 до 100% соответственно).
Анализ присоединения различных фрагментов к циклу C2N4O2 показывает, что, например, структура 6 (ЭО = 4645.6 кДж/кг) образуется в результате присоединения к циклу C2N4O2 фрагмента R2 = N2O. Присоединение кислорода к фрагменту N2O с образованием фрагмента NONO (структура 4) приводит к уменьшению энтальпии на 393 кДж/кг (от 4645.6 до 4252.6 кДж/кг). Однако замена N2O на N3H (структура 8, ЭО = = 4815.7 кДж/кг) приводит к увеличению энтальпии на 170 кДж/кг. Структура 8, в которой к циклу C2N4O2 присоединен фрагмент N3H, имеет энтальпию образования 4815.7 кДж/кг. Однако замена водорода на фрагмент NO2 (структура 5) приводит к уменьшению энтальпии на 420 кДж/кг подобно тенденции, отмеченной для структур 3 и 1. Увеличение цепочки NONO (структура 4) до N4O2 (структура 7) приводит к увеличению энтальпии на 503 кДж/кг (от 4252.6 до 4755.2 кДж/кг).
Для экспериментально изученных веществ C2N6O4 (структура 4), C2N6O3 (структура 6), C2N8O4 (структура 7) рассчитанные значения энтальпии ${{\Delta }_{f}}H_{{298}}^{^\circ }\left( {\text{г}} \right)$ больше экспериментальных на 8–15%, что значительно меньше разброса экспериментальных значений для этих соединений (табл. 2).
Таким образом, можно отметить, что энтальпия для C2N4O2-R2 (структуры 4–8) растет в ряду R2 = NONO, N4O2, NON, N2ON2O, N3H с увеличением массовой доли азота и уменьшением массовой доли кислорода. При одинаковом соотношении азота и кислорода энтальпия выше при образовании шестичленного цикла по сравнению с пятичленным, образованными за счет четырех (структура 7) и трех (структура 5) атомов азота группы R2 соответственно.
Следует отметить, что влияние заместителей R2 на энтальпию структур 4–8 не превышает 563 кДж/кг, что составляет 12–13% от общей величины, в то время как сшивка пятичленных колец C2N2O фрагментом R1 = N2 (структура 10) увеличивает энтальпию почти вдвое по сравнению с NH (структура 3). Однако почти такой же эффект оказывает образование из структуры 3 бирадикала (триплета) со структурой 9 за счет удаления атомов водорода.
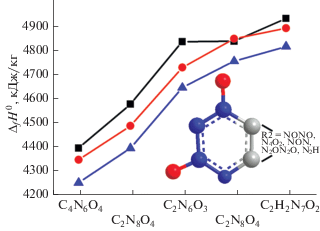
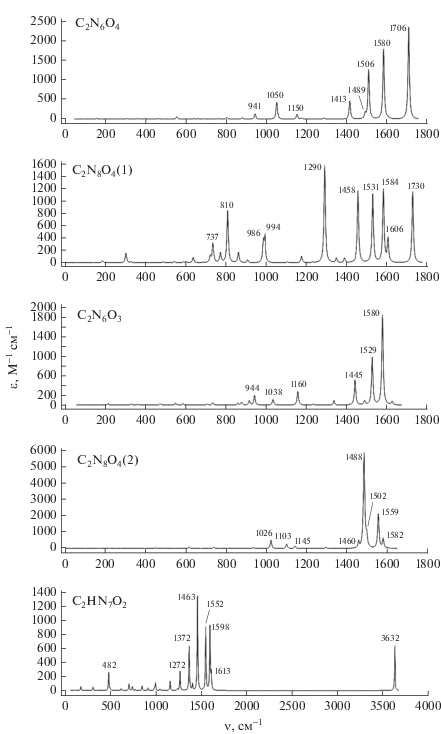
ИК-спектры. На рис. 4 и 5 приведены ИК-спектры поглощения рассчитанных молекул (в соответствии с рис. 2 и 3). Смещения атомов для наиболее интенсивных колебаний приведены на рис. 6 и 7.
Согласно рис. 4 и 6, можно сказать, что наиболее высокие частоты с заметной интенсивностью имеет фрагмент R1: ~1700 (R1 = NO2, структура 1), 3631 (R1 = H, структура 3), 1612 см–1 (R1 = N2, структура 10). Вторые по величине – частоты, отвечающие колебаниям связи C–C: 1542 (C4N8O6), 1639, 1480, 1340 (C4H2N6O2), 1186 см–1 (C4N8O2). Далее с гораздо меньшей интенсивностью идут более низкие частоты, отвечающие колебаниям фрагмента N2O.
Анализируя рис. 5 и 7, можно отметить, что наиболее высокое значение частоты с заметной интенсивностью имеет фрагмент NO2 в структуре 5 (1730 см–1). Вторые по величине – частоты, отвечающие колебаниям фрагмента NO (1458–1706 см–1). Далее с гораздо меньшей интенсивностью идут более низкие частоты, отвечающие колебаниям связей C–C (1445–1613 см–1) и N–N (941–1272 см–1).
ЗАКЛЮЧЕНИЕ
Методами квантово-химических расчетов на уровне B3LYP/6-311+G(2d,p) и комбинированных методов G4(MP2) и G4 с использованием прикладного пакета GAUSSIAN 09 на высокопроизводительных вычислительных ресурсах были проведены оценки термохимических свойств ряда газообразных веществ (в кДж/кг): C4N8O6 (2854.0), C4HN7O4 (2932.3), C4H2N6O2 (3104.8), C2N6O4 (4347.3), C2N8O4 (4252.6), C2N6O3 (4645.6), C2N8O4 (4755.2), C2HN7O2 (4815.7), C4N6O2 (5153.2), C4N8O2 (5609.1). Для экспериментально изученных веществ C2N6O4, C2N6O3, C2N8O4 рассчитанные на уровне G4 значения энтальпии ${{\Delta }_{f}}H_{{298}}^{^\circ }\left( {\text{г}} \right)$ больше экспериментальных на 8–15%, что значительно меньше опубликованной разницы экспериментальных значений для этих соединений. Термохимические данные для высокоэнергетических соединений C4N8O6, C4HN7O4, C4H2N6O2, C2N8O4, C2HN7O2, C4N6O2 и C4N8O2, а также ИК-спектры поглощения, структурные параметры и смещения атомов для наиболее интенсивных колебаний впервые получены в настоящей работе. Эти данные могут быть использованы как справочные и позволяют выделить наиболее перспективные группы веществ для работы в качестве высокоэнергетических компонент перспективного топлива.
Показано, что энтальпия для C2N4O2–R2 растет в ряду R2 = NONO, N4O2, NON, N2ON2O, N3H с ростом массовой доли азота и уменьшением массовой доли кислорода. При одинаковом соотношении азота и кислорода энтальпия выше при образовании шестичленного цикла по сравнению с пятичленным, образованных четырьмя и тремя атомами азота группы R2 соответственно.
Продемонстрировано, что использование расчета G4(MP2) для выбранного класса молекул дает очень близкие (в пределах 1–3%) по сравнению с расчетом G4 результаты и ведет к существенной (в 3–8 раз) экономии расчетного времени. Однако результаты, полученные на уровне B3LYP/6-311+G(2d,p), отличаются от наиболее точных, полученных на уровне G4, на 2–11%.
В работе продемонстрирована возможность компьютерного дизайна гипотетически перспективных, еще не синтезированных молекул, что открывает новые возможности для эффективного отбора перспективных для исследования молекул.
Список литературы
Hosseini S.G., Moeini K., Abdelbaky M.S.M., Garcia-Granda S. // J. Struct. Chem. 2020. V. 61. № 3. P. 389. https://doi.org/10.26902/JSC_id52850
Абдулов Х.Ш., Муллоев Н.У., Табаров С.Х., Ходиев М.Х. // J. Struct. Chem. 2020. V. 61. № 4. P. 540. https://doi.org/10.26902/JSC_id53992
Lv G., Zhang D.-L., Wang D. et al. // J. Struct. Chem. 2019. V. 60. № 7. P. 1219. https://doi.org/10.26902/JSC_id43057
Curtiss L.A., Redfern P.C., Raghavachari K. // J. Chem. Phys. 2007. V. 126. P. 084108. https://doi.org/10.1063/1.2436888
Karton A., Sylvetsky N., Martin J.M.L. // J. Comput. Chem. 2017. V. 38. P. 2063.
Сунцова М.А. Прогнозирование энтальпий образования новых азотсодержащих высокоэнергетических соединений на основе квантово-химических расчетов. Дис. … канд. хим. наук. М., 2016.
Kiselev V.G., Gritsan N.P., Zarko V.E. et al. // Combust. Explos. ShockWaves. 2007. V. 43. P. 562. https://doi.org/10.1007/s10573-007-0074-6
Muthurajian H., Sivabalan R., Talavar M.B. et al. // J. Hazard. Mater. 2006. V. 133. P. 30.
Ryce B.M., Hai S.V., Hare J. // Combust. Flame. 1999. V. 118. P. 445.
Byrd E.F.C., Ryce B.M. // J. Phys. Chem. A. 2006. V. 110. P. 1005.
Mikhailov O.V., Chachkov D.V. // Russ. J. Inorg. Chem. 2020. V. 65. № 5. P. 646. [Михайлов О.В., Чачков Д.В. // Журн. неорган. химии. 2020. Т. 65. № 5. С. 598.]https://doi.org/10.1134/S0036023620050174
Ma D.X., Xia Q.Y. // J. Struct. Chem. 2018. V. 59. № 1. P. 28. https://doi.org/10.1134/S0022476618010055
Hasan T., Ghalib R.M., Mehdi S.H. et al. // J. Struct. Chem. 2018. V. 59. № 5. P. 1078. https://doi.org/10.1134/S0022476618050098
Zeyrek C.T., Unver H., Temiz-Arpaci O. et al. // J. Struct. Chem. 2019. V. 60. № 2. P. 241. https://doi.org/10.1134/S0022476619020094
Turovtsev V.V., Orlov Yu.D., Kaplunov I.A. // J. Struct. Chem. 2018. V. 59. № 8. P. 1960. [Туровцев В.В., Орлов Ю.Д., Каплунов И.А. // Журн. структур. химии. 2018. Т. 59. № 8. С. 2021.]https://doi.org/10.1134/S0022476618080279
Curtiss L.A., Raghavachari K., Redfern P.C. et al. // J. Chem. Phys. 1998. V. 109. P. 7764. https://doi.org/10.1063/1.477422
Curtiss L.A., Raghavachari K., Redfern P.C. et al. // Chem. Phys. Lett. 1999. V. 314. P. 101. https://doi.org/10.1016/S0009-2614(99)01126-4
Baboul A.G., Curtiss L.A., Redfern P.C., Raghavachari K. // J. Chem. Phys. 1999. V. 110. P. 7650. https://doi.org/10.1063/1.478676
Belisario-Lara D., Mebel A.M., Kaiser R.I. // J. Phys. Chem. A. 2018. V. 122. № 16. P. 3980. https://doi.org/10.1021/acs.jpca.8b01836
Morozov A.N., Mebel A.M., Kaiser R.I. // J. Phys. Chem. A. 2018. V. 122. № 22. P. 4920. https://doi.org/10.1021/acs.jpca.8b02934
Zhao L., Yang T., Kaiser R.I. et al. // Phys. Chem. Chem. Phys. 2017. V. 19. P. 15780. https://doi.org/10.1039/C7CP01571B
Zhang F.T., Kaiser R.I., Kislov V.V. et al. // J. Phys. Chem. Lett. 2011. V. 2. № 14. P. 1731. https://doi.org/10.1021/jz200715u
Zhang F.T., Kaiser R.I., Golan A. et al. // J. Phys. Chem. A. 2012. V. 116. P. 3541. https://doi.org/10.1021/jp300875s
Kaiser R.I., Belau L., Leone S.R. et al. // Chem. Phys. Chem. 2007. V. 8. P. 1236. https://doi.org/10.1002/cphc.200700109
Kaiser R.I., Mebel A., Kostko O., Ahmed M. // Chem. Phys. Lett. 2010. V. 485. P. 281. https://doi.org/10.1016/j.cplett.2009.12.027
Kaiser R.I., Maksyutenko P., Ennis C. et al. // Faraday Discuss. 2010. V. 147. P. 429. https://doi.org/10.1039/C003599H
Kaiser R.I., Sun B.J., Lin H.M. et al. // Astrophys. J. 2010. V. 719. P. 1884. https://doi.org/10.1088/0004-637X/719/2/1884
Kostko O., Zhou J., Sun B.J. et al. // Astrophys. J. 2010. V. 717. P. 674. https://doi.org/10.1088/0004-637X/717/2/674
Kaiser R.I., Krishtal S.P., Mebel A.M. et al. // Astrophys. J. 2012. V. 761. P. 178. https://doi.org/10.1088/0004-637X/761/2/178
Golan A., Ahmed M., Mebel A.M., Kaiser R.I. // Phys. Chem. Chem. Phys. 2013. V. 15. P. 341. https://doi.org/10.1039/C2CP42848B
Curtiss L.A., Redfern P.C., Raghavachari K. // Comput. Mol. Sci. 2011. V. 1. P. 810. https://doi.org/10.1002/wcms.59
Curtiss L.A., Raghavachari K., Redfern P.C., Pople J.A. // J. Chem. Phys. 1997. V. 106. № 3. P. 1063. https://doi.org/10.1063/1.473182
NIST-JANAF Thermochemical tables https://janaf.nist.gov/ [Электронный ресурс: дата доступа 08.08.2019].
Computational Chemistry Comparison and Benchmark DataBase https://cccbdb.nist.gov/hf0k.asp [Электронный ресурс: дата доступа 08.08.2019].
Ефимов А.И., Белорукова Л.П., Василькова И.В. и др. Свойства неорганических соединений. Справочник. Л.: Химия, 1983.
Гурвич Л.В. Энергии разрыва химических связей. М.: Наука, 1974.
Frisch M.J., Trucks G.W., Schlegel H.B. et al. Gaussian 09, Revision B.01. Gaussian, Inc., Wallingford CT, 2010.
Curtiss L.A., Redfern P.C., Raghavachari K. // J. Chem. Phys. 2005. V. 123. 124107. https://doi.org/10.1063/1.2039080
He X., Zhang J., Gao H. // Int. J. Quant. Chem. 2012. V. 112. P. 1688. https://doi.org/10.1002/qua.23163
Волохов В.М., Зюбина Т.С., Волохов А.В. и др. // Химическая физика. 2020. № 12 (в печати).
Becke A.D. // J. Chem. Phys. 1993. V. 98. № 4. P. 5648. https://doi.org/10.1063/1.464906
Johnson B.J., Gill P.M.W., Pople J.A. // J. Chem. Phys. 1993. V. 98. № 4. P. 5612. https://doi.org/10.1063/1.464906
Voevodin Vl., Antonov A., Nikitenko D. et al. // Supercomputing Frontiers and Innovations, 2019. V. 6. P. 4. https://doi.org/10.14529/jsfi190201
Воеводин В.В., Жуматий С.А., Соболев С.И. и др. // Открытые системы. СУБД. 2012. № 7. С. 36.
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии