

Журнал неорганической химии, 2021, T. 66, № 1, стр. 41-48
Гидротермально-микроволновой синтез α-МоО3
З. А. Фаттахова a, Э. Г. Вовкотруб b, Г. С. Захарова a, *
a Институт химии твердого тела УрО РАН
620990 Екатеринбург, ул. Первомайская, 91, Россия
b Институт высокотемпературной электрохимии УрО РАН
620990 Екатеринбург, ул. Академическая, 20, Россия
* E-mail: volkov@ihim.uran.ru
Поступила в редакцию 07.07.2020
После доработки 20.08.2020
Принята к публикации 25.08.2020
Аннотация
Гидротермально-микроволновой обработкой водного раствора пероксомолибденовой и щавелевой кислот, взятых в молярном соотношении Мо : Н2С2О4 · 2H2O = 1 : (0.25‒1), синтезирован триоксид молибдена орторомбической сингонии α-МоО3. Структурные особенности формирования α-МоО3 изучены с помощью рентгенофазового анализа, КР-спектроскопии и пикнометрического метода определения плотности. Методами сканирующей электронной микроскопии и низкотемпературной адсорбции азота определено влияние условий синтеза на морфологию и текстурные характеристики порошков триоксида молибдена. Установлено, что молярное соотношение компонентов реакционной массы является определяющим фактором процесса получения МоО3. Из спектров оптического поглощения определена ширина запрещенной зоны синтезированных образцов.
ВВЕДЕНИЕ
Триоксид молибдена является полупроводником n-типа с шириной запрещенной зоны 2.9−3.6 эВ, зависящей от размера частиц и упорядоченности кристаллической решетки [1]. MoO3 находит применение в катализе [2], химических источниках тока [3], газосенсорных устройствах [4, 5], а также используется как электродно-активный материал для определения рН и концентрации ионов щелочных металлов в растворе [6]. Существует пять полиморфных модификаций МоО3: орторомбическая фаза α-МоО3 (пр. гр. Pbnm), моноклинная фаза высокого давления МоО3-II (P21/m), метастабильные гексагональная фаза h-МоО3 (P63/m) и моноклинные фазы β-МоО3 (P21/c), β'-МоО3 (P21/n) [7]. Широкое применение орторомбической модификации триоксида молибдена обусловлено ее уникальной слоистой структурой и высокой термической стабильностью.
Существует целый ряд методов получения α-MoO3. Классическим методом синтеза является пиролиз молибденсодержащих соединений, выполняющих роль прекурсора. В качестве прекурсора могут быть использованы оксид диэтилдитиокарбамат молибдена Mo((C2H5)2NCS2)2O2 [8], тетратиомолибдат аммония (NH4)2MoS4 [9], формиат молибдена НСООМо4О12ОН [10]. α-MoO3 также может быть получен термической обработкой на воздухе диоксида молибдена [11, 12], h-MoO3 [13], тетрагидрата парамолибдата аммония (NH4)6Mo7O24 · 4H2O [14]. Несмотря на простоту, метод термического разложения молибденсодержащих соединений имеет ограниченное применение, связанное со сложностью контролирования гранулометрического состава конечного продукта. Одним из наиболее универсальных методов синтеза нанокристаллического триоксида молибдена является гидротермальный метод (ГТ), который позволяет надежно управлять размером частиц и их морфологией [15]. В качестве источника молибдена может быть использован металлический молибден [16], ацетилацетонат молибдена MoO2(С5Н7О2)2 [17], молибдат натрия Na2MoO4 [18], молибдат аммония (NH4)2MoO4 [19] и т.д. В зависимости от условий синтеза и типа используемого прекурсора частицы MoO3 формируются в виде нанолент [20–23], наностержней [24–26], нанолистов и наноцветов [27]. Морфологию МоО3 также можно контролировать введением различных солей и добавок, таких как нитрат калия KNO3, нитрат натрия NaNO3, нитрат кальция Ca(NO3)2, нитрат лантана La(NO3)3 [28], хлорид хрома CrCl3 · 6H2O [29], поливинилпирролидон (C6H9NO)n [30, 31], цетилтриметиламмония бромид [Me(CH2)15NMe3]Br (ЦТАБ) [32]. В последнее время исследователи стали все чаще применять одну из модификаций ГТ-метода, в которой нагрев реакционной массы производится посредством микроволнового воздействия [33]. Большое развитие гидротермально-микроволновой синтез (ГТ-МВ) получил благодаря высокой скорости протекания реакции, равномерному нагреву всего объема реакционной среды, использованию перемешивания, обеспечивающего полную гомогенизацию реакционной массы [34]. ГТ-МВ обработка смеси (NH4)6Mo7O24 · 4H2O, HNO3 и ЦТАБ позволяет синтезировать наностержни МоО3 длиной 10−12 мкм и диаметром 50 нм [35]. Наноленты МоО3 длиной 5−8 мкм и шириной 100−300 нм могут быть получены ГТ-МВ обработкой смеси молибденовой кислоты Н2МоО4, пероксида водорода, азотной кислоты [36]. МоО3 в виде наноремней длиной несколько мкм и шириной 50−150 нм может быть получен ГТ-МВ обработкой смеси пероксомолибденовой и муравьиной кислот [37].
В настоящей работе впервые ГТ-МВ обработкой водного раствора пероксомолибденовой и щавелевой кислот синтезирован триоксид молибдена орторомбической сингонии, исследована его морфология, текстурные и оптические свойства.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В качестве исходных веществ использовали порошок металлического молибдена (99.9 мас. % Мо), 30%-ный раствор пероксида водорода марки “ос. ч.”, щавелевую кислоту Н2С2О4 · 2H2O марки “х. ч.”. Синтез МоО3 проводили в одну стадию. Порошок молибдена растворяли при охлаждении (5‒10°С) в Н2О2 с образованием желтого раствора пероксомолибденовой кислоты. К полученному раствору добавляли Н2С2О4 · 2H2O в молярном соотношении Мо : Н2С2О4 · 2H2O = 1 : (0.25‒2). Затем реакционный раствор помещали в гидротермально-микроволновой реактор Monowave 300 (Anton Parr) и выдерживали при температуре 140−220°С в течение 20 мин. Синтез вели при избыточном давлении 10−12 бар и постоянном перемешивании со скоростью 300 мин–1. Полученные осадки голубого цвета отфильтровывали, промывали водой и сушили на воздухе при комнатной температуре. Продукты синтеза обозначены как МоО3-T, где T − температура синтеза.
Рентгенофазовый анализ (РФА) образцов проводили на дифрактометре XRD-7000 (Shimadzu) (CuKα-излучение, λ = 1.5418 Å). Спектры комбинационного рассеяния света регистрировали на спектрометре U1000 (Renishaw) с использованием твердотельного лазера LCM-S-111 с длиной волны 532 нм и мощностью 40 мВт. Исследование морфологии образцов выполняли на сканирующем электронном микроскопе (СЭМ) JSM 6390 LA (Jeol). Текстурные характеристики (удельная поверхность, пористость) образцов определяли анализатором Gemini VII (Micromeritics) по низкотемпературной адсорбции азота. Пробоподготовку образцов проводили вакуумированием при 200°С в течение 2 ч. На основании полученных изотерм сорбции азота рассчитывали удельную поверхность по методу Брунауэра‒Эммета‒Теллера. Анализ пористости материалов выполняли с использованием данных изотерм сорбции по методу Баррета‒Джойнера‒Халенды. Пикнометрическую плотность порошков МоО3-Т определяли с помощью гелиевого пикнометра AccuPyc II 1340 (Micromeritics) с камерой объемом 1 см3. Заполнение ячейки материалом составляло 50‒60%. Точность измерения массы образцов составляла ±0.0001 г. Для корректности данных выполняли 10 циклов измерения плотности для каждого образца. Погрешность измерения составляла ±0.01 г/см3. Спектры поглощения в УФ-, видимом и ближнем ИК-диапазонах регистрировали на спектрометре UV-2600 (Shimadzu) в диапазоне длин волн 190‒1400 нм. Оптическую ширину запрещенной зоны определяли графически с использованием формулы:
где α ‒ коэффициент поглощения, hν ‒ энергия фотона, Eg ‒ оптическая ширина запрещенной зоны, А ‒ постоянная, не зависящая от частоты ν, n – показатель, зависящий от типа оптического перехода, равный 1/2, 3/2, 2 и 3 для прямых разрешенных, прямых запрещенных, непрямых разрешенных и непрямых запрещенных переходов соответственно. Для определения оптической ширины запрещенной зоны (Eg) были построены зависимости (αhν)1/n = f(hν), прямолинейные участки которых экстраполировали до пересечения с осью энергии фотонов [38]. Наилучшее совпадение с линейностью на указанных зависимостях при переменных значениях n указывает на наличие определенного типа межзонного перехода. Погрешность определения составляла ±0.01 эВ.РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Согласно данным РФА (рис. 1), синтезированные оксиды МоО3-T кристаллизуются в орторомбической сингонии (пр. гр. Pbnm) с параметрами элементарной ячейки, представленными в табл. 1. На дифрактограммах образцов наблюдаются интенсивные дифракционные пики семейства (0k0), свидетельствующие о преобладании анизотропного роста частиц МоО3 вдоль оси b. Увеличение температуры синтеза триоксида молибдена сопровождается уменьшением параметров a и c, при этом параметр b растет.
Рис. 1.
Дифрактограммы порошков МоО3-T при T = = 140 (1), 180 (2), 220°С (3) и позиции брегговских пиков α-МоО3 по данным JCPDS 5-508.
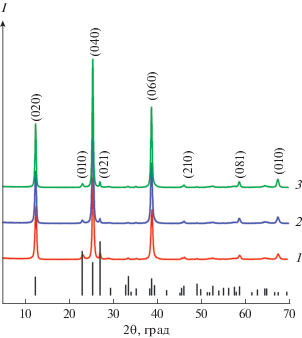
Таблица 1.
Параметры элементарной ячейки и результаты определения рентгеновской и пикнометрической плотности МоO3-T
| Образец | Параметры элементарной ячейки | ρр | ρп | Δρ | n × 1021, см–3 | |||
|---|---|---|---|---|---|---|---|---|
| a, Å | b, Å | c, Å | V, Å3 | г/см3 | ||||
| МоО3-140 | 3.991(8) | 13.843(9) | 3.700(8) | 204.5(2) | 4.69 | 5.64 | 0.95 | 3.9 |
| МоО3-180 | 3.959(8) | 13.870(2) | 3.694(9) | 202.3(9) | 4.71 | 4.89 | 0.18 | 0.8 |
| МоО3-220 | 3.960(2) | 13.875(0) | 3.683(2) | 202.3(8) | 4.72 | 4.88 | 0.16 | 0.7 |
| α-МоО3 (JCPDS 5-508) | 3.962 | 13.858 | 3.697 | 202.99 | 4.71 | – | – | – |
С использованием уравнения Шеррера была проведена оценка среднего размера кристаллитов МоO3-T:
где D – размер кристаллитов, k – безразмерный коэффициент формы частиц, равный 0.93 (постоянная Шеррера), Δ(2θ) – ширина пика на половине высоты, θ – брэгговский угол, λ – длина волны рентгеновского излучения. Расчет вели по интенсивным и хорошо разрешенным дифракционным пикам (020), (040), (060). Средний размер кристаллитов МоO3-140, МоO3-180, МоO3-220 составляет 19, 21 и 27 нм соответственно. Очевидно, что увеличение температуры ГТ-МВ обработки способствует формированию более крупных частиц МоО3. Следует отметить, что щавелевая кислота, благодаря наличию карбонильной функциональной группы C=O, выполняет роль мягкого восстанавливающего агента, способствуя формированию молибден-кислородных цепочек в результате реакции поликонденсации за счет ионов Мо5+ [39].Для оценки дефектности структуры синтезированных МоО3-T была определены рентгеновская и пикнометрическая плотность образцов. Рентгенографическую плотность (ρр) рассчитывали по уравнению [40]:
где z – число формульных единиц в элементарной ячейке, М – молярная масса (г/моль), NA – число Авогадро (6.022 × 1023 моль–1), V – объем элементарной ячейки (см3). Результаты расчетов рентгеновской и экспериментально определенной пикнометрической плотности (ρп), а также разница их значений (Δρ) приведены в табл. 1. Как видно из полученных результатов, рентгеновская и пикнометрическая плотность материалов различается. С увеличением температуры синтеза Δρ уменьшается и для МоО3-220 составляет 0.16 г/см3. Наибольшее различие плотности наблюдается для образца MoO3-140 (Δρ = 0.95 г/см3). Такое влияние температуры синтеза на пикнометрическую плотность MoO3-T, по-видимому, обусловлено несовершенством кристаллической решетки оксида, вызванным образованием различных типов дефектов. Концентрацию дефектов (n) оценивали с помощью уравнения [41]:Наименьшая концентрация дефектов наблюдается в образцах, синтезированных при температурах 180–220°С. Очевидно, что повышение температуры синтеза сопровождается упорядочением кристаллической решетки α-МоО3, что приводит к заметному уменьшению концентрации дефектов. Полученные данные хорошо согласуются и с уменьшением объема кристаллической решетки образцов MoO3-T, полученных при более высоких температурах.
Согласно СЭМ-исследованиям, синтезированные порошки МоО3-T имеют морфологию частиц, подобную агломерированным ремням, ширина которых увеличивается с ростом температуры синтеза и равна 100−190, 180−250, 185−520 нм для МоО3-140, МоО3-180, МоО3-220 соответственно (рис. 2). Длина частиц практически не зависит от условий получения и составляет несколько мкм.

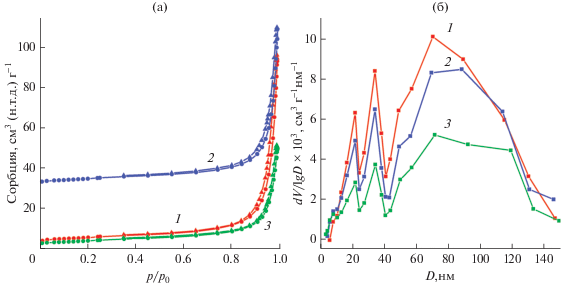
На рис. 3 представлены результаты исследования удельной поверхности и пористости синтезированных образцов. Согласно классификации ИЮПАК [42], изотермы сорбции МоО3-T относятся к IV типу с петлей гистерезиса Н3 (рис. 3а). С повышением температуры синтеза величина удельной поверхности уменьшается и для образцов МоО3-140, МоО3-180, МоО3-220 составляет 19.4, 17.7, 14.4 м2/г соответственно. По-видимому, это связано с укрупнением частиц в процессе синтеза, что хорошо согласуется с данными РФА. Распределение пор по размерам в синтезированных образцах МоО3-T является полимодальным, о чем свидетельствует ход кривой распределения пор по размерам с характеристическими максимумами в области существования мезо- и микропор (рис. 3б). Значение диаметра пор первых двух экстремумов, соответствующее преимущественному их размеру, одинаково для всех образцов МоО3-T независимо от температуры синтеза и составляет 22 и 34 нм соответственно. Третий экстремум на кривой распределения указывает на наличие макропор в образцах, преимущественный размер которых с ростом температуры синтеза изменяется от 70 до 93 нм. Увеличение размеров макропор в ряду МоО3-140, МоО3-180, МоО3-220 также сопровождается уменьшением объема пор, что дополнительно с ростом размера частиц при повышении температуры синтеза приводит к уменьшению удельной поверхности.
Рис. 3.
Изотермы сорбции (а) и кривые распределения пор по размерам (б) порошков МоО3-T при T = 140 (1), 180 (2), 220°С (3).
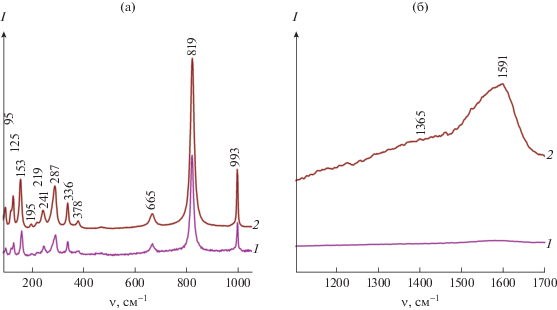
Методом КР-спектроскопии на примере образца МоО3-160 изучено влияние соотношения компонентов реакционной смеси на структуру и фазовый состав продуктов реакции. Независимо от содержания щавелевой кислоты в реакционной массе в спектрах КР в интервале 90‒1050 см–1 фиксируются вибрационные моды, характерные для α-МоО3 (рис. 4а) [43]. Наблюдаемые пики при 95, 125 и 153 см–1 соответствуют колебаниям кристаллической решетки оксида. Полосы с максимумами при 195, 219 и 241 см–1 относятся к деформационным ножничным колебаниям связей О–Мо–О. Деформационное веерное колебание связи О=Мо=О проявляется при 287 см–1. Деформационные колебания связи О–Мо–О фиксируются при 336 и 378 см–1. Валентные колебания мостиковых связей О–Мо–О проявляются при 665 см–1. Интенсивные узкие полосы при 819 и 993 см–1 относятся к валентным колебаниям кратных связей Мо=О. Дополнительный анализ спектров КР в высокочастотной области позволил идентифицировать присутствие углерода. При молярном соотношении Мо : Н2С2О4 · 2H2O = 1 : 2 в спектре КР МоО3-160 наблюдается широкий размытый пик в интервале 1250‒1700 см–1, представляющий собой суперпозицию двух пиков с максимумами при 1365 и 1591 см–1 (рис. 4б). Пик при 1365 см–1 описывает D-линию, которая соответствует колебаниям атомов углерода с sp3-типом гибридизации и свидетельствует о наличии разупорядочения углеродной составляющей. Пик при 1591 см–1 описывает G-линию, соответствующую колебаниям атомов углерода в sp2-гибридизации, и указывает на наличие в образце мелкокристаллического графита [44]. Таким образом, исследования, выполненные с использованием КР-спектроскопии, показали, что варьированием молярного соотношения компонентов реакционной массы может быть получен α-МоО3 и композит на его основе МоО3/С.
Рис. 4.
Спектры КР порошков МоО3-160 при молярном соотношении Мо : Н2С2О4 · 2H2O = 1 : 1 (1) и 1 : 2 (2) в диапазоне частот 90‒1050 см–1 (а) и 1200‒1700 см–1 (б).
На рис. 5 приведены спектры поглощения синтезированных образцов в сравнении с коммерческим триоксидом молибдена. Спектры всех образцов МоО3-T демонстрируют интенсивную полосу поглощения с максимумом при λ ~ 300 нм, описывающую перенос заряда из валентной зоны О2р в зону проводимости Мо4d (O2p → Mo4d) в октаэдрах [MoO6] [45] (рис. 5а). Измерения спектральных характеристик в УФ- и видимой областях спектра позволили определить край полосы собственного поглощения МоО3, формирующегося прямыми разрешенными переходами, и рассчитать оптическую ширину запрещенной зоны (рис. 5б). Установлено, что оптическая ширина запрещенной зоны коммерческого α-МоО3 и синтезированных ГТ-МВ методом образцов МоО3-140, МоО3-180, МоО3-220 составляет 3.04 и 3.09, 3.13, 3.18 эВ соответственно. Значение ширины запрещенной зоны коммерческого α-МоО3, размер частиц которых составляет несколько мкм [46], значительно меньше по сравнению с Eg для MoO3-T. Согласно данным [47], уменьшение ширины запрещенной зоны с увеличением размера частиц вызвано квантово-размерным эффектом. Оптическая ширина запрещенной зоны для образцов МоО3-T, синтезированных при различных температурах, определяется несколькими факторами. Во-первых, квантово-размерным эффектом, приводящим к снижению Eg для частиц, полученных при повышенных температурах и отличающихся более крупными размерами. С другой стороны, уменьшение концентрации структурных дефектов, сопровождаемое упорядочением и совершенством кристаллической решетки α-МоО3 с ростом температуры синтеза, обусловливает значительный рост ширины запрещенной зоны [48]. Результатом суммарного эффекта является увеличение Eg для образцов в ряду МоО3-140, МоО3-180, МоО3-220.
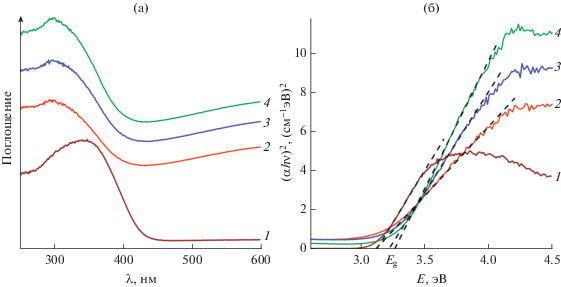
ЗАКЛЮЧЕНИЕ
Гидротермально-микроволновой обработкой водного раствора пероксомолибденовой и щавелевой кислот синтезирован триоксид молибдена орторомбической сингонии с морфологией частиц, подобной ремням. Изучены морфологические особенности, текстурные и оптические свойства образцов в зависимости от условий синтеза. Увеличение температуры синтеза приводит к образованию более крупных частиц α-МоО3, формирование которых сопровождается уменьшением дефектности кристаллической структуры и удельной поверхности. Установлено влияние размера частиц и наличия структурных дефектов на ширину запрещенной зоны. Определены условия получения композита на основе триоксида молибдена МоО3/С, подробное изучение физико-химических и практических свойств которого является предметом дальнейших исследований.
Список литературы
Sinaim H., Ham D.J., Lee J.S. et al. // J. Alloys Compd. 2012. V. 516. P. 172. https://doi.org/10.1016/j.jallcom.2011.12.024
Chithambararaj A., Sanjini N.S., Velmathi S. et al. // Phys. Chem. Chem. Phys. 2013. V. 15. № 35. P. 14 761. https://doi.org/10.1039/c3cp51796a
Hu X., Zhang W., Liu X. et al. // Chem. Soc. Rev. 2015. V. 44. № 8. P. 2376. https://doi.org/10.1039/c4cs00350k
Mandal B., Aaryashree, Das M. et al. // Mater. Res. Bull. 2019. V. 109. P. 281. https://doi.org/10.1016/j.materresbull.2018.09.041
Liu Y., Yang S., Lu Y. et al. // Appl. Surf. Sci. 2015. V. 359. P. 114. https://doi.org/10.1016/j.apsusc.2015.10.071
Zakharova G.S., Fattakhova Z.A., Zhu Q. et al. // J. Electroanal. Chem. 2019. V. 840. P. 187. https://doi.org/10.1016/j.jelechem.2019.03.072
Lunk H.-J., Hartl H. // ChemTexts. 2017. V. 3. P. 1. https://doi.org/10.1007/s40828-017-0048-6
Bai H.X., Liu X.H., Zhang Y.C. // Mater. Lett. 2009. V. 63. № 1. P. 100. https://doi.org/10.1016/j.matlet.2008.09.016
Гараев А.М., Рзаева А., Бабаева Н. // Вестн. Томского гос. ун-та. Сер. Химия. 2016. Т. 5. № 3. С. 49.
Троицкая И.Б., Гаврилова Т.А., Костровский В.Г. и др. // Фундаментальные проблемы современного материаловедения. 2009. Т. 6. № 1. С. 64.
Sui L., Xu Y.-M., Zhanget X.-F. et al. // Sens. Actuators, B. 2015. V. 208. P. 406. https://doi.org/10.1016/j.snb.2014.10.138
Jiang W., Wei D., Zhang S. et al. // New J. Chem. 2018. V. 42. P. 15111. https://doi.org/10.1039/c8nj03539c
Huang J., Yan J., Li J. et al. // J. Alloys Compd. 2016. V. 688. P. 588. https://doi.org/10.1016/j.jallcom.2016.07.055
He S., Li W., Feng L. et al. // J. Alloys Compd. 2019. V. 783. P. 574. https://doi.org/10.1016/j.jallcom.2018.12.349
Симонено Т.Л., Бочарова В.А., Симоненко Н.П. и др. // Журн. неорган. химии. 2020. Т. 65. № 4. С. 435. https://doi.org/10.31857/S0044457X20040182
Xiao X., Peng Z., Chen C. et al. // NanoEnergy. 2014. V. 9. P. 355. https://doi.org/10.1016/j.nanoen.2014.08.001
Wang Z., Madhavi S., Lou X.W. (David) // J. Phys. Chem. C. 2012. V. 116. № 23. P. 12508. https://doi.org/10.1021/jp304216z
Camacho-Bragado G.A., Jose-Yacaman M. // Appl. Phys. A. 2006. V. 82. № 1. P. 19. https://doi.org/10.1007/s00339-005-3370-6
Li Y., Ren H., Jiao Q. // Ferroelectrics. 2018. V. 527. № 1. P. 113. https://doi.org/10.1080/00150193.2018.1450556
Куа Е., Пу Я., Хао Ю. и др. // Электрохимия. 2015. Т. 51. № 2. С. 145. https://doi.org/10.7668/S0424857015020097
Zhou L., Yang L., Yuan P. et al. // J. Phys. Chem. C. 2010. V. 114. № 49. P. 21868. https://doi.org/10.1021/jp108778v
He S., Li W., Feng L. et al. // J. Alloys Compd. 2019. V. 783. P. 574. https://doi.org/10.1016/j.jallcom.2018.12.349
Song Y., Zhao J., Zhao Y. et al. // CrystEngComm. 2016. V. 18. № 34. P. 6502. https://doi.org/10.1039/c6ce01090c
Wang Q., Sun J., Wang Q. et al. // J. Mater. Chem. A. 2015. V. 3. № 9. P. 5083. https://doi.org/10.1039/c5ta00127g
Khandare L., Terdale S.S., Late D.J. // Adv. Device Mater. 2016. V. 2. № 2. P. 15. https://doi.org/10.1080/20550308.2016.1221605
Волков В.Л., Захарова Г.С., Кузнецов М.В. // Журн. неорган. химии. 2008. Т. 53. № 11. С. 1807. [Volkov V.L., Zakharova G.S., Kuznetsov M.V. // Russ. J. Inorg. Chem. 2008. V. 53. № 11. P. 1686. https://doi.org/10.1134/S003602360811003X]
Li G., Jiang L., Pang S. et al. // J. Phys. Chem. B. 2006. V. 110. № 48. P. 24472.
Xia T., Li Q., Liu X. et al. // J. Phys. Chem. B. 2006. V. 110. № 5. P. 2006. https://doi.org/10.1021/jp055945n
Li Y. // Physica E. 2017. V. 94. P. 22. https://doi.org/10.1016/j.physe.2017.07.010
Ji H., Zeng W., Li Y. // J. Mater. Sci. Mater. Electron. 2019. https://doi.org/10.1007/s10854-019-00967-0
Gong J.W., Wan X.F. // Mater. Technol. 2015. V. 30. № 6. P. 332. https://doi.org/10.1179/1753555715Y.0000000008
Wang S., Zhang Y., Ma X. et al. // Solid State Commun. 2005. V. 136. № 5. P. 283. https://doi.org/10.1016/j.ssc.2005.08.022
Фаттахова З.А., Захарова Г.С. // Журн. неорган. химии. 2020. Т. 65. № 4. С. 458. https://doi.org/10.31857/S0044457X20040054
Иванова О.С., Теплоногова М.А., Япрынцев А.Д. и др. // Журн. неорган. химии. 2018. Т. 63. № 6. С. 678. [Ivanova O.S., Yapryntsev A.D., Baranchikov A.E. et al. // Russ. J. Inorg. Chem. 2018. Т. 63. № 6. P. 708. https://doi.org/10.1134/S0036023618060128https://doi.org/10.7868/S0044457X1806003X
Phuruangrat A., Ham D.J., Thongtem S. et al. // Electrochem. Commun. 2009. V. 11. № 9. P. 1740. https://doi.org/10.1016/j.elecom.2009.07.005
Wang L., Zhang X., Ma Y. et al. // Mater. Lett. 2016. V. 164. P. 623. https://doi.org/10.1016/j.matlet.2015.11.076
Zakharova G.S., Schmidt C., Ottmann A. et al. // J. Solid State Electrochem. 2019. V. 22. P. 3651. https://doi.org/10.1007/s10008-018-4073-1
Tauc J. The optical properties of solids. Academic. Waltham. 1966.
Zakharova G.S., Taschner C., Volkov V.L. et al. // Solid State Sci. 2007. V. 9. № 11. P. 1028. https://doi.org/10.1016/j.solidstatesciences.2007.07.022
Шаскольская М.П. Кристаллография. М.: Высш. шк., 1984.
Суриков В.И., Суриков В.И., Семенюк Н.А. и др. // Изв. вузов. Физика. 2017. Т. 60. № 10. С. 153.
Sing K.S.W., Everett D.H., Haul R.A.W. et al. // Pure Appl. Chem. 1985. V. 57. № 4. P. 603. https://doi.org/10.1351/pac198557040603
Phuruangrat A., Chen J.S., Lou X.W. et al. // Appl. Phys. A. 2012. V. 107. P. 249. https://doi.org/10.1007/s00339-012-6771-3
Ferrari A.C., Robertson J. // Phys. Rev. B. 2000. V. 61. № 20. P. 14095. https://doi.org/10.1103/physrevb.61.14095
Itoh M., Hayakawa K., Oishi S. // J. Phys. Condens. 2001. V. 13. № 31. P. 6853. https://doi.org/10.1088/0953-8984/13/31/319
Zakharova G.S., Podval’naya N.V. // J. Anal. Chem. 2013. V. 68. № 1. P. 50. https://doi.org/10.1134/S1061934813010140
Alivisatos A.P. // J. Phys. Chem. 1996. V. 100. № 31. P. 13 226. https://doi.org/10.1021/jp9535506
Ghasemi B., Hajakbari F., Hajakbari A. // Inorg. Nano-Met. Chem. 2020. V. 50. № 5. P. 414. https://doi.org/10.1080/24701556.2020.1716008
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии