

Журнал неорганической химии, 2020, T. 65, № 8, стр. 1119-1127
Синтез и искровое плазменное спекание микрокристаллического диоксида тория для изделий топливного назначения
О. О. Шичалин a, b, *, К. Р. Фролов b, И. Ю. Буравлев a, b, И. Г. Тананаев b, c, d, В. В. Фаизова b, С. А. Азон a, b, Н. И. Андреева c, Е. К. Папынов a, b
a Институт химии ДВО РАН
690022 Владивосток, пр-т 100-летия Владивостока, 159, Россия
b Дальневосточный федеральный университет
690090 Владивосток, ул. Суханова, 8, Россия
c Озерский технологический институт – филиал НИЯУ МИФИ
456780 Озёрск, пр-т Победы, 48, Россия
d Институт физической химии и электрохимии РАН им. А.Н. Фрумкина
119991 Москва, Ленинский пр-т, 31, корп. 4, Россия
* E-mail: oleg_shich@mail.ru
Поступила в редакцию 04.03.2020
После доработки 20.03.2020
Принята к публикации 27.03.2020
Аннотация
Диоксид тория представляет будущую перспективу для атомных реакторов четвертого поколения. В этой связи проблемы технологий выделения тория из облученного ядерного топлива, его перевод в топливное сырье и фабрикация топливных изделий требуемого качества нуждаются в эффективных решениях. В работе проведен синтез микрокристаллического мезопористого порошка ThO2 через осаждение оксалатного комплекса из нитратного раствора и его консолидация по технологии искрового плазменного спекания (ИПС). Изучено влияние температуры ИПС в интервале 1000–1600°С на динамику усадки порошка ThO2, фазовый состав, микроструктуру и плотность керамических изделий таблеточного типа. Определены параметры микротвердости образцов по Виккерсу. Показано, что высокоскоростная ИПС-консолидация порошка при 1600°С и давлении 80 МПа позволяет получать ThO2-керамику с относительной плотностью 95.2%. При этом в качестве исходного сырья может быть использован микрокристаллический порошок с дисперсностью 3–4 мкм взамен менее доступного наноразмерного сырья. Показана целесообразность изучения ИПС для его возможной промышленной адаптации.
ВВЕДЕНИЕ
Диоксид тория (ThO2) является основой ядерного топливного цикла, реализуемого на изотопе Th-232 [1], который при захвате тепловых нейтронов образует делящийся нуклид U-233. Современная атомная энергетика уделяет особое внимание ториевому ядерному топливу и позиционирует его как альтернативу для замены U-235. Это связано с большей доступностью тория, его лучшими теплофизическими свойствами, высокой температурой плавления, повышенной теплопроводностью и низким коэффициентом теплового расширения. Относительно инертный ThO2 имеет повышенную устойчивость к окислению и радиационному воздействию по сравнению с диоксидом урана (UO2) [2]. При выработке такого топлива образуется меньшее количество трансурановых элементов, что соответствует режиму нераспространения ядерного оружия при использовании ThO2 в традиционных легководных реакторах [3].
Перспективы использования ядерного ThO2-топлива экспериментально изучаются на реакторах китайского (HTR-10), канадского (CANDU), индийского (AHWR) производства, а также реакторах HTGR на гелиевом охлаждении [3–5]. Опыт показывает, что наряду с очевидными преимуществами такого топлива имеются и серьезные недостатки. В частности, проблема заключается в высоком гамма-излучении дочернего изотопа U-232, образующегося при распаде Th-232. Кроме того, на стадии переработки облученного ядерного топлива отмечается низкая способность ThO2 к растворению в азотной кислоте, что является обязательной процедурой, в результате усложняются традиционные технологии его выделения из отходов и фабрикация порошков регенератов в топливное изделие требуемого качества.
В виду необходимости наработки ThO2 из нитратной системы как основного сырьевого компонента для топлива в промышленности ориентируются на различные технологические способы. В частности, активно развивается синтез дисперсного ThO2 переводом нитрата тория в оксалат тория Th(C2O4)2 · 2H2O осадительным синтезом с последующей кальцинацией осадка [6, 7]. Метод осаждения оксалата удобен в технологическом применении для переработки и разделения ядерных отходов [8] вследствие своей универсальности, так как позволяет формировать оксалаты с одним катионом, например Th(IV) или U(IV), а также с двумя и более катионами, например Th(IV) + U(IV) [6, 9]. Однако важным фактором остается выбор условий отжига оксалатных солей с целью получения чистого и пригодного для топливных изделий оксидного сырья, а также его последующего спекания. Сложность заключается в механизме превращения оксалата в оксид при термодеструкции. При низкой температуре происходит дегидратация, при более высокой температуре оксалатная группа разлагается на CO, CO2, карбонаты или оксокарбонаты металла в зависимости от газовой среды. В каждом отдельном случае присутствует вероятность содержания примесей в оксидном порошке, а также возможно изменение дисперсности и пористости его частиц, что сказывается на качестве конечного топлива и может усложнять технологический процесс его производства. В результате комплексного исследования в работе [7] описано влияние путей осаждения оксалата, его измельчения и гомогенизации (смешения), а также температуры прокаливания порошков ThO2 на основе оксалата, включая размер и пористость зерна, на эффективность спекания и качество топливных таблеток. В литературе отмечается значительное влияние негативных факторов на традиционные способы консолидации порошка, но применение нестандартных решений позволяет снизить их воздействие на режимы изготовления и качество изделий. В частности, известна современная технология искрового плазменного спекания (ИПС), перспективы которой уже доказаны для широкого ряда керамических систем в работах [10–15], в том числе и для UO2 [16–18]. Уникальные возможности ИПС для консолидации ThO2 рассмотрены только в нескольких работах [19–21], где было реализовано спекание коммерческих порошков ThO2 керамического сорта заданной чистоты и дисперсности, а также получены смешанные торий-урановые системы. Формирование керамики на основе порошка ThO2 с применением ИПС, синтезированного из оксалата Th(C2O4)2 · 2H2O, было изучено только в работе [22], где исследователи успешно реализовали спекание образцов с использованием порошков ThO2 двух видов: синтезированного по методике [7] и коммерческого, после чего провели сравнение характеристик плотности конечных изделий. Несмотря на высокую значимость достигнутых результатов, обосновавших влияние размера зерна на эффективность спекания и конечные характеристики изделия, в указанном исследовании использовали только наноразмерные порошки ThO2 (13, 19 и 34 нм), синтез которых довольно сложный и дорогостоящий. При этом единственным варьируемым параметром процесса ИПС была скорость разогрева (50, 100 и 200 град/мин), температурный режим был неизменным (1600°С), и его выбор не обсуждался.
В связи с этим цель настоящей работы – синтез и последующее искровое плазменное спекание микрокристаллического порошка ThO2, полученного из оксалатного комплекса, а также исследование состава, структуры и плотности топливных таблеток в зависимости от температуры спекания.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Синтез дисперсного ThO2 проводили прямым осаждением Th(IV) из азотнокислого раствора при добавлении 0.1–0.5 М раствора оксалата натрия. Образующийся осадок оксалата тория Th(C2O4)2 · 6H2O центрифугировали (режимы) и отделяли от маточного раствора декантированием. Полученный осадок Th(C2O4)2 · 6H2O прокаливали в муфельной печи Nabertherm GmbH (Германия) при 700°C в течение 5 ч на воздухе.
Химическая реакция осаждения оксалата тория:
(1)
$\begin{gathered} {\text{T}}{{{\text{h}}}^{{ + 4}}} + 2{{{\text{H}}}_{{\text{2}}}}{{{\text{C}}}_{{\text{2}}}}{{{\text{O}}}_{4}} + 6{{{\text{H}}}_{{\text{2}}}}{\text{O}} \to \\ \to \,\,{\text{Th}}{{\left( {{{{\text{C}}}_{{\text{2}}}}{{{\text{O}}}_{{\text{4}}}}} \right)}_{{\text{2}}}} \cdot 2{{{\text{H}}}_{{\text{2}}}}{\text{O}} + 4{{{\text{H}}}_{{\text{3}}}}{{{\text{O}}}^{ + }}. \\ \end{gathered} $Синтез керамического ThO2. Консолидацию синтезированного порошка ThO2 с получением керамического компакта таблеточного типа проводили искровым плазменным спеканием на установке LABOX-625 (Sinter Land Incorporation LTD, Япония) по следующей схеме. Навеску 0.5 г порошка ThO2 с фракцией 100–500 мкм помещали в графитовую пресс-форму (внешний диаметр 30 мм, внутренний диаметр 8 мм, высота 30 мм), подпрессовывали (давление 20.7 МПа), затем заготовку помещали в вакуумную камеру (давление 6 Па) и спекали. Для предотвращения припекания консолидируемого порошка к пресс-форме и плунжерам и легкого извлечения полученного компаунда использовали графитовую фольгу толщиной 200 мкм. Пресс-форму оборачивали в теплоизолирующую ткань для снижения теплопотерь при разогреве. Температура спекания составляла 1000, 1200, 1400 и 1600°C, скорость разогрева регулировалась стадиями: 300 град/мин в диапазоне температур от 0 до 650°C и 100 град/мин от 650°C и выше. Образец выдерживали при максимальной температуре 5 мин, затем охлаждали до комнатной температуры в течение 30 мин. Давление прессования было постоянным в течение всего процесса и составляло 75 МПа. Разогрев спекаемого материала осуществляли импульсным током в режиме On/Off с периодичностью подачи/пауз импульсов 12/2, т.е. длительность импульсов 39.6 мс и пауз между ними 6.6 мс.
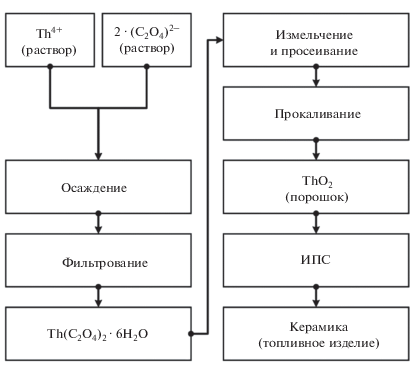
Общая схема синтеза образцов представлена на рис. 1.
Гранулометрический состав порошка ThO2 определяли методом лазерной дифракции на анализаторе частиц Analysette-22 NanoTec/MicroTec/XT Fritsch (Германия). Идентификацию кристаллических фаз в составе образцов проводили с помощью рентгенофазового анализа (РФА) (CuKα-излучение, Ni-фильтр, λсредн = 1.5418 Å, диапазон углов съемки 10°–80°, шаг сканирования 0.02°, скорость регистрации спектров 5 град/мин) на многоцелевом рентгеновском дифрактометре D8 Advance Bruker AXS (Германия). База данных РФА Powder Diffraction FileTM (Soorya N Kabekkodu, 2007). Удельную поверхность исходного порошка и керамических образцов определяли на анализаторе низкотемпературной адсорбции азота при 77 K на приборе Autosorb IQ фирмы Quantochrome (США), для расчета применяли модель БЭТ и DFT. Изображения структуры исследуемых материалов были получены методом растровой электронной микроскопии (РЭМ) на приборе Carl Zeiss Ultra 55 (Германия) c катодом на полевой эмиссии при ускоряющем напряжении 1–5 кВ и токе пучка I ≈ 100 pA. Микротвердость по методу Виккерса (HV) измеряли при нагрузке HV0.5 с помощью микротвердомера HMV-G-FA-D Shimadzu (Япония). Плотность образцов определяли методом гидростатического взвешивания на весах AdventurerTM OHAUS Corporation (США).
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
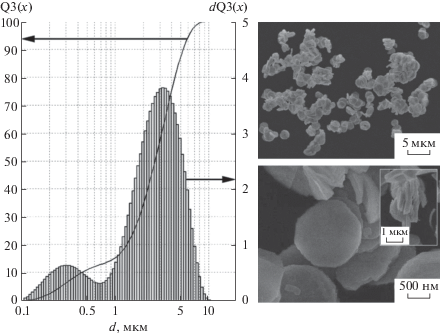
Согласно данным гранулометрического состава (рис. 2), метод прямого осаждения Th(C2O4)2 · 6H2O с последующей кальцинацией осадка до ThO2 обеспечивает получение порошка с дисперсностью частиц 0.1–10 мкм. Средний размер фракции составляет 3–4 мкм. РЭМ-изображения подтверждают установленную дисперсность и отражают слоистую структуру частиц (рис. 2).
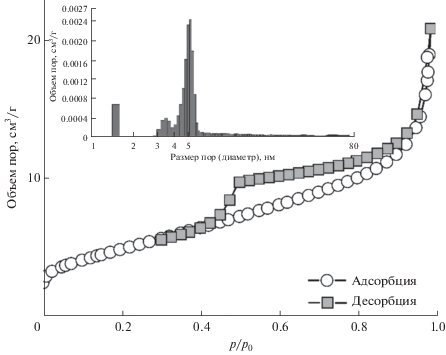
По результатам структурного анализа, синтезированный порошок ThO2 имеет мезопористую структуру, что отражено на изотерме сорбции–десорбции азота (рис. 3), средний размер пор составляет 3–6 нм (рис. 3, врезка). Величина удельной площади поверхности (Sуд) достигает 17 м2/г.
Рис. 3.
Изотерма адсорбции–десорбции азота и гистограмма распределения диаметра пор (врезка) для микрокристаллического порошка ThO2.
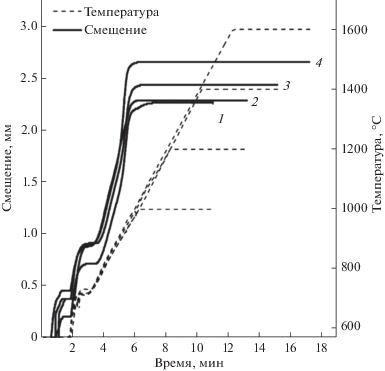
Динамика уплотнения порошка ThO2 описывается дилатометрическими зависимостями (рис. 4). На кривых отмечены три стадии усадки порошка. Две начальные стадии протекают в первые 3 мин процесса при температурах разогрева до 800–850°С, что вызвано деструкцией, перегруппировкой и упаковкой частиц за счет механического прессования. Последняя стадия возникает на 3–6 мин цикла при разогреве свыше 900°С, что обусловлено процессами пластической деформации, вязкого течения и зернограничной диффузии в твердой фазе при термическом воздействии. Величина усадки материала повышается с ростом температуры ИПС.
Рис. 4.
Дилатометрические зависимости уплотнения (усадки) порошка ThO2 в условиях ИПС при температуре: 1000 (1), 1200 (2), 1400 (3), 1600°С (4).
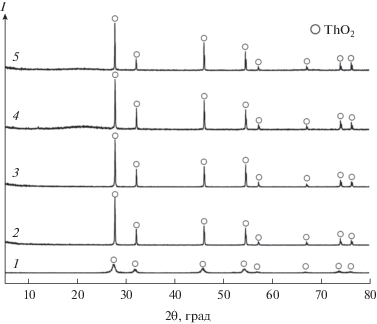
По данным РФА (рис. 5), фазовый состав образцов керамики ThO2 отличается большей степенью кристалличности по сравнению с исходным порошком, что обусловлено активным ростом кристаллитов при спекании. В составе образцов отсутствуют посторонние примеси и не образуются дополнительные фазы.
Рис. 5.
Дифрактограммы образцов ThO2: исходный порошок (1) и образцы керамики на его основе, полученные ИПС при 1000 (2), 1200 (3), 1400 (4), 1600°С (5).
Изменения микроструктуры образцов при различных температурных режимах ИПС отражены на металлографических и РЭМ-снимках поверхности продольных срезов керамики (рис. 6). Выявлено, что при низких температурах спекания формируется керамика с большим количеством пор, сформированных упаковкой частиц и равномерно распределенных по всей исследуемой поверхности образцов. Повышение температуры приводит к активному росту частиц, увеличению спекаемой площади их контактов и, соответственно, к постепенному снижению общего количества пор.
Рис. 6.
Металлографические снимки (а–г) и РЭМ-изображения (а*–г*) поверхности срезов образцов керамики на основе ThO2, полученных ИПС при разной температуре.

Установлено, что величина плотности образцов ThO2-керамики повышается линейно с ростом температуры спекания (табл. 1). Наибольшее значение теоретической плотности (95.2%) достигается при температуре 1600°С, что обусловлено снижением пористости образца за счет активного роста зерна (рис. 6) и наибольшей величиной усадки порошка при спекании (рис. 4).
Таблица 1.
Характеристики образцов ThO2-керамики, полученных ИПС
| Образец | tсинт, °С | pпрес, МПа | ρэксп, г/см3 | ρотн, % | Sуд (БЭТ), м2/г | HV |
|---|---|---|---|---|---|---|
| ThO2-1 | 1000 | 80 | 9.0793 | 90.8 | 0.4 | 598 |
| ThO2-2 | 1200 | 9.0907 | 90.9 | 0.3 | 537 | |
| ThO2-3 | 1400 | 9.1003 | 91.0 | 0.1 | 418 | |
| ThO2-4 | 1600 | 9.5295 | 95.2 | 0.1 | 336 |
Выявлено, что величина микротвердости (HV) образцов снижается с ростом температуры (табл. 1), это связано со значительной агломерацией мелких пор в крупные в объеме керамики в ходе роста зерна при спекании. Объемные дефекты в структуре керамики образуются при высокоскоростном нагреве в результате слияния линейных и поверхностных дефектов межзеренного пространства из-за неравномерности распределения тепла. Как следствие, из-за заданной порами структуры керамики ThO2 микропластической неустойчивости вдавливание индентора в образец облегчается (рис. 7). По отпечаткам индентора, представленным на рис. 7, отчетливо прослеживается разница в качестве поверхности образцов керамики. При увеличении нагрузки индентора при вдавливании в испытуемый образец с HV0.2 до HV2 отпечаток имеет структуру поверхности керамики с наиболее высоким значением микротвердости (598 HV).

ЗАКЛЮЧЕНИЕ
Реализован и описан полный цикл процесса получения образцов топливных изделий на основе диоксида тория. Синтезирован микрокристаллический (средний размер зерна 3–4 мкм) мезопористый (Sуд = 17 м2/г) порошок ThO2 путем прямого осаждения оксалатного комплекса металла из нитратного раствора c последующей его кальцинацией. Проведена высокоскоростная консолидация полученного порошка по технологии ИПС и получены образцы плотной ThO2-керамики. По дилатометрическим зависимостям исследовано влияние ИПС-разогрева в интервале 1000–1600°С на динамику спекания и установлено, что усадка порошка протекает в несколько стадий при разной температуре разогрева и вызвана механическим и термическим воздействием. Методами РФА, РЭМ и металлографии установлена стабильность фазового состава образцов керамики и выявлены их структурные изменения, связанные с ростом зерна и исключением пористости, при повышении температуры ИПС. Определено, что относительная плотность полученных образцов в пределах 95.2% достигается при 1600°С за 17 мин одного цикла спекания. Установлено, что микротвердость керамики снижается (598–336 HV) с ростом температуры из-за значительной агломерации мелких пор в крупные дефекты в межзеренном пространстве. Показано, что изготовление изделий топливного назначения эффективно реализуется по технологии ИПС при более привлекательных технологических режимах с использованием микрокристаллических порошков взамен менее доступного наноразмерного сырья.
Список литературы
Ault T., Krahn S., Croff A. // Ann. Nucl. Energy. 2017. V. 110. P. 726. https://doi.org/10.1016/j.anucene.2017.06.026
Humphrey U.E., Khandaker M.U. // Renew. Sustain. Energy Rev. 2018. V. 97. P. 259. https://doi.org/10.1016/j.rser.2018.08.019
Schaffer M.B. // Energy Policy. 2013. V. 60. P. 4. https://doi.org/10.1016/j.enpol.2013.04.062
Anantharaman K., Shivakumar V., Saha D. // J. Nucl. Mater. 2008. V. 383. P. 119. https://doi.org/10.1016/j.jnucmat.2008.08.042
Wojtaszek D., Colton A.V., Bromley B.P. et al. // Ann. Nucl. Energy. 2018. V. 111. P. 152. https://doi.org/10.1016/j.anucene.2017.09.004
Tyrpekl V., Vigier J.F., Manara D. et al. // J. Nucl. Mater. 2015. V. 460. P. 200. https://doi.org/10.1016/j.jnucmat.2015.02.027
Wangle T., Tyrpekl V., Cagno S. et al. // J. Nucl. Mater. 2017. V. 495. P. 128. https://doi.org/10.1016/j.jnucmat.2017.07.046
Abraham F., Arab-Chapelet B., Rivenet M. et al. // Coord. Chem. Rev. 2014. V. 266–267. P. 28. https://doi.org/10.1016/j.ccr.2013.08.036
Clavier N., Hingant N., Rivenet M. et al. // Inorg. Chem. 2010. V. 49. P. 1921. https://doi.org/10.1021/ic902343r
Simonenko N.P., Simonenko E.P. // Ceram. Int. 2018. V. 44. P. 19879. https://doi.org/10.1016/j.ceramint.2018.07.249
Simonenko T.L., Kalinina M.V., Simonenko N.P. et al. // Int. J. Hydrogen Energy. 2019. V. 44. P. 20345. https://doi.org/10.1016/j.ijhydene.2019.05.231
Tokita M. // Spark Plasma Sintering (SPS) Method, Systems, and Applications / Ed. Somiya S. Handb. Adv. Ceram. Mater. Appl. Process. Prop. Elsevier Inc., 2013. P. 1149. https://doi.org/10.1016/B978-012654640-8/50007-9
Simonenko E.P., Simonenko N.P., Gordeev A.N. et al. // Russ. J. Inorg. Chem. 2018. V. 63 P. 421. https://doi.org/10.1134/S0036023618040186
Papynov E.K., Shichalin O.O., Buravlev I.Y. et al. // Russ. J. Inorg. Chem. 2020. V. 65. P. 263. https://doi.org/10.1134/S0036023620020138
Papynov E.K., Shichalin O.O., Medkov M.A. et al. // Glas. Phys. Chem. 2018. V. 44. P. 632. https://doi.org/10.1134/S1087659618060159
Ge L., Subhash G., Baney R.H. et al. // J. Nucl. Mater. 2013. V. 435 P. 1. https://doi.org/10.1016/j.jnucmat.2012.12.010
Papynov E.K., Shichalin O.O., Mironenko A.Y. et al. // Radiochemistry. 2018. V. 60. P. 362. https://doi.org/10.1134/S1066362218040045
Chen Z., Subhash G., Tulenko J.S. // J. Nucl. Mater. 2014. V. 454. P. 427. https://doi.org/10.1016/j.jnucmat.2014.08.023
Malakkal L., Prasad A., Ranasinghe J. et al. // J. Nucl. Mater. 2019. V. 527. P. 1. https://doi.org/10.1016/j.jnucmat.2019.151811
Saoudi M., Staicu D., Mouris J. et al. // J. Nucl. Mater. 2018. V. 500. P. 381. https://doi.org/10.1016/j.jnucmat.2018.01.014
Muta H., Murakami Y., Uno M. et al. // J. Nucl. Sci. Technol. 2013. V. 50. P. 181. https://doi.org/10.1080/00223131.2013.757468
Tyrpekl V., Cologna M., Robba D., Somers J. // J. Eur. Ceram. Soc. 2016. V. 36. P. 767. https://doi.org/10.1016/j.jeurceramsoc.2015.11.006
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии