

Журнал неорганической химии, 2020, T. 65, № 7, стр. 922-930
Каталитическая активность октаметоксизамещенного тетрафенилпорфирината кобальта(II) в реакции окисления тетратерпенов пероксидом водорода
О. Р. Симонова a, *, С. А. Зданович a, С. В. Зайцева a, О. И. Койфман a, b
a Институт химии растворов им. Г.А. Крестова РАН
153045 Иваново, ул. Академическая, 1, Россия
b Ивановский государственный химико-технологический университет
153000 Иваново, Шереметьевский пр-т, 7, Россия
* E-mail: ors@isc-ras.ru
Поступила в редакцию 31.12.2019
После доработки 03.02.2020
Принята к публикации 27.02.2020
Аннотация
Спектральными методами исследована каталитическая активность порфирината кобальта(II) в реакции окислительного разложения β-каротина и ликопина пероксидом водорода. Получены кинетические характеристики редокс-процесса, предложен его возможный механизм. Показана зависимость каталитической активности от состава координационного центра и наличия дополнительных редокс-активаторов. Установлено, что окислительной деструкции тетратерпенов предшествует координация на катионе металла комплекса пероксида водорода с его последующей активацией. В результате этой активации образуется высокореакционный интермедиат с радикалом, локализованным на метоксигруппе, способный инициировать свободнорадикальное окисление.
ВВЕДЕНИЕ
Разбалансировка состояния про- и антиоксидантных систем организма приводит к нарушениям текущего уровня и взаимопревращениям активных форм кислорода (АФК) и свободных радикалов. Такой сбой в окислительном метаболизме вызывает активацию процессов липопероксидации с последующим молекулярным повреждением мембран и генетического аппарата клетки и, как следствие, возникновение тяжелых заболеваний. Для предотвращения или корректировки этого процесса необходимо купировать генерацию АФК. Одним из способов редуцировать гиперсинтез АФК является введение в организм синтетических аналогов внутриклеточных ферментов и низкомолекулярных антиоксидантов.
Моделирование природных ферментов связано с поиском и созданием систем, проявляющих высокую каталитическую активность. Исследования последних лет показывают, что в качестве биомиметических катализаторов успешно применяются макроциклические комплексы биологически активных металлов [1–13]. В работе [12] демонстрируется высокая деполимеризационная и каталитическая активность порфиринатов марганца в реакциях окисления органических ароматических полютантов в мягких условиях. Антибактериальную активность в отношении G+- и G–-бактерий и антиоксидантные свойства проявляет мезо-тетра(4-метоксифенил)порфирин циркония(IV) и его аксиальные комплексы с электроноакцепторным лигандом. Авторы [13] отмечают, что высокая активность комплексов циркония связана с эффектом сочетания в одной молекуле двух редокс-активаторов в виде катиона металла и порфиринового лиганда.
Структурное разнообразие порфиринов и их аналогов определяет широкий диапазон свойств этих соединений, а значит, дает возможность управлять активностью биомиметической системы посредством увеличения реакционных центров за счет модификации периферии и внутренней координационной сферы. Биомиметические исследования с применением синтетических моделей каталаз и пероксидаз на основе этих соединений свидетельствуют о том, что условием проявления ферментоподобной активности является образование высоковалентных оксокомплексов или высокореакционных интермедиатов с редокс-активаторами на периферии за счет активации пероксида и разрыва в нем связи О–О [14–23]. Это стимулирует интерес к изучению каталитических свойств макроциклических тетрапиррольных соединений и объясняет выбор в качестве объекта исследования 5,10,15,20-тетракис(2,5-диметоксифенил)порфината кобальта(II). Систематические исследования влияния модификации структуры макроциклического лиганда и окружения реакционного центра комплексов на их каталитические свойства в редокс-процессах необходимы для детального понимания активации свободнорадикального окисления.
Как известно, антиоксидантная система состоит из ферментативных и неферментативных звеньев. Одними из низкомолекулярных антиоксидантов являются каротиноиды, которые участвуют во второй линии защиты, избавляя клетку от избытка биорадикалов.
Считается, что каротиноиды с высокосопряженной системой двойных связей являются превосходными объектами для свободнорадикальной атаки, в результате которой образуются сложные интермедиаты [24–27]. С одной стороны, они могут быть малоактивными, а с другой – участвовать в редокс-превращениях, генерируя активные радикалы, и выступать в качестве прооксидантов. Окисление каротиноидов – сложный и до конца не изученный процесс, поэтому исследования окислительных реакций с участием каротиноидов и тестирование их как антиоксидантов остаются актуальными.
В настоящей работе в качестве модели одной из стадий многокомпонентной антиоксидантной защиты исследована реакция окисления тетратерпенов (β-каротина и ликопина) пероксидом водорода (Н2О2) в присутствии порфирината кобальта(II) в ацетонитриле при 295 K.
CoIITPP(OCH3)8
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
5,10,15,20-тетракис(2,5-диметоксифенил)порфинат кобальта(II) (CoTPP(OCH3)8) (1) синтезировали по методике [28]. 5,10,15,20-тетракис(2,5-диметоксифенил)порфин (29.0 мг, 0.04 ммоль) и ацетат кобальта(II) Co(AcO)2 ⋅ 4H2O (47.0 мг, 0.19 ммоль) растворяли в ацетонитриле и кипятили в атмосфере инертного газа в течение 60 мин. Окончание реакции определяли по прекращению изменений в УФ-видимой части электронного спектра поглощения (ЭСП) комплекса. После упаривания растворителя под вакуумом и промывки осадка водой комплекс хроматографировали на оксиде алюминия (элюент – хлороформ) и сушили под вакуумом. Выход 5,10,15,20-тетракис(2,5-диметоксифенил)порфината кобальта(II) 95%. ЭСП Co(II)TPP(OCH3)8 (ацетонитрил (λmax, нм (lg ε)): 413 (5.14), 528 (4.16). ИК-спектр Co(II)TPP(OCH3)8 (НПВО, ацетонитрил), см–1: 3041–2973 ν(C–H.), 1653–1378 (колебания порфиринового и бензольных циклов), 1271–1263 ν(CPh–О–С), 1168 ν(C–О, –ОСН3), 1042 ν(CPh–О–С), 729–655 ν(C–N), 457 ν(Co–Np). MALDI-TOF: m/z = 911.86 для [C52H44N4O8Co]+.
Электронные спектры поглощения регистрировали на приборе Varian Cary 50 в диапазоне 350–800 нм при 295 K с использованием кварцевых кювет толщиной 1 см. ИК-спектры получены методом НПВО (нарушенного полного внутреннего отражения) на спектрометре Bruker Vertex V80 с помощью приставки Harrick MVP2 SeriesTM (материал призмы – алмаз) в диапазоне частот 4000–400 см–1 (по 64 сканирования в среднем) с разрешением 2 см–1 при стандартной температуре в ацетонитриле.
Масс-спектры (MALDI-TOF) регистрировали на времяпролетном масс-спектрометре с матрично-ассоциированной лазерной десорбцией Axima Confidence.
В работе использовали пероксид водорода (30%), ацетонитрил (99.8%), каротин (97%) и ликопин (Ph. s. s.) производства Sigma-Aldrich.
Кинетические параметры реакции получены с использованием методики [29]. Эффективные константы скорости (kэф) и константы скорости реакции окисления каротина (kV) при 295 K определены с учетом изменения оптической плотности (A) реакционной смеси путем оптимизации зависимостей lg(A0 – A∞)/(Aτ – A∞) – τ и lg kэф–${\text{lg}}C_{{{\text{тетратерп}}}}^{0}$ (A0, Aτ и A∞ – оптическая плотность на рабочих длинах волн при λmax = 480 и 473 нм (для каротина и ликопина) в начальный, текущий момент времени и в конце реакции соответственно) при постоянной концентрации исходного соединения и пероксида водорода (${{C}_{{{{{\text{Н}}}_{{\text{2}}}}{{{\text{О}}}_{{\text{2}}}}}}}$ = 1.4 моль/л) и различных концентрациях каротина (Ccar = 2.3 × 10–4–5.9 × 10–4 моль/л) и ликопина (Cлик = 4.9 × 10–5–1.35 × 10–4 моль/л). Порядок по тетратерпену устанавливали по тангенсу угла наклона линейной зависимости lgkэф–${\text{lg}}C_{{{\text{тетратерп}}}}^{0}.$ Оптимизацию проводили с использованием метода наименьших квадратов.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Каротиноиды по механизму действия являются тушителями активных форм кислорода и оказывают профилактическое действие в отношении целого ряда заболеваний, которые связаны с нарушением уровня пероксидного окисления липидов клеточных мембран и других свободнорадикальных процессов в организме. Самым активным из группы каротиноидов является ликопин. Данные по влиянию β-каротина на окисление биологически активных субстратов имеют противоречивый характер, поскольку наряду с антиокислительным действием обнаружены и проокислительные эффекты. Считается, что антиокислительное и антирадикальное действие β-каротина проявляется только при малых парциальных давлениях кислорода. В связи с этим реакции окисления β-каротина под действием свободных радикалов активно изучаются как в водной среде, так и в органических растворителях. Время окислительного разложения β-каротина сильно зависит от природы окислителя и среды [24, 30–32].
Каталитическая активность порфиринатов металлов в окислении каротиноидов пероксидами недостаточно изучена и представлена в единичных работах. Авторами [19, 23, 25, 32] исследованы реакции окислительного разложения каротина третбутилгидропероксидом в гексане и мета-хлорнадбензойной кислотой в бензоле в присутствии тетрадихлорофенилпорфирин рутения(II) и карбонил(5,10,15,20-тетра-2,4,6-триметилфенилпорфирин) рутения(II) в качестве соокислителей. Установлено, что порфиринаты рутения вступают в реакцию в активной оксоформе O=Ru(IV). В работах [19, 23] показано, что активной окислительной частицей в системе димерный комплекс железа(IV)–трет-бутилгидропероксид–каротиноид является оксоформа O=FeP+•=C=FeP (Р = TАP, Рс). Поэтому следует ожидать, что при активации пероксида водорода порфиринатом кобальта(II) образуется окисленная форма комплекса, способная участвовать в разложении тетратерпенов.
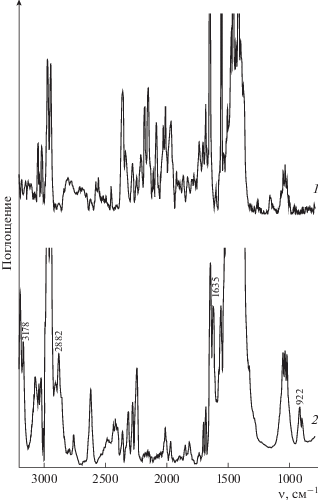
Добавление пероксида водорода (${{C}_{{{{{\text{Н}}}_{{\text{2}}}}{{{\text{О}}}_{{\text{2}}}}}}}$ = = 1.4 моль/л) к раствору порфирината кобальта(II) в ацетонитриле (${{C}_{{{\text{CoTPP}}{{{\left( {{\text{OC}}{{{\text{H}}}_{{\text{3}}}}} \right)}}_{{\text{8}}}}}}}$ = 5.4 × 10–6 моль/л) приводит к изменению ЭСП комплекса. Наблюдается батохромное смещение полос при 413 и 528 нм на 20 нм с одновременным появлением полос при 500 и 619 нм (рис. 1). Подобная трансформация ЭСП (λmax = 433, 500, 548 и 619 нм) свидетельствует об образовании двуокисленной формы исходного комплекса с радикалом на макроцикле. Механизм формирования высокоокисленных форм порфиринатов металлов под действием пероксидов подробно описан в литературе [10, 19, 21–23, 27, 33–37]. Следует отметить, что через минуту в реакционной смеси визуально наблюдается выделение кислорода. В ЭСП этот факт отражается нарушением монотонности спектральной кривой в результате возникновения низкоинтенсивных хаотических дискретных флуктуаций. Образование двуокисленной формы комплекса кобальта подтверждается методами ИК-спектроскопии и масс-спектрометрии. В ИК-спектре реакционной смеси после добавления Н2О2 (${{C}_{{{{{\text{Н}}}_{{\text{2}}}}{{{\text{О}}}_{{\text{2}}}}}}}$ = = 1.0 моль/л) имеют место полосы колебаний ν(С–Н) = 3178, 2882 см–1 в $-{\text{ОСН}}_{2}^{\centerdot }$ и полоса при 1635 см–1, соответствующая колебаниям связей C=O в хинонах [38, 39]. Наблюдается также полоса ν(О–О) с частотой 922 см–1 [40], характерная для пероксогруппы, но не в составе пероксида водорода [41–43] (рис. 2).
Рис. 1.
Изменение ЭСП каротина (Ccar = 4.86 × 10–4 моль/л) в реакции с пероксидом водорода (${{C}_{{{{{\text{H}}}_{{\text{2}}}}{{{\text{O}}}_{{\text{2}}}}}}}$ = 1.4 моль/л) в присутствии CoIITPP(OCH3)8 (${{С}_{{{\text{C}}{{{\text{o}}}^{{{\text{II}}}}}{\text{TPP}}{{{\left( {{\text{OC}}{{{\text{H}}}_{{\text{3}}}}} \right)}}_{{\text{8}}}}}}}$ = 5.4 × 10–6 моль/л) в ацетонитриле при 295 K (а); изменение ЭСП CoIITPP(OCH3)8 при активации пероксида водорода (б).
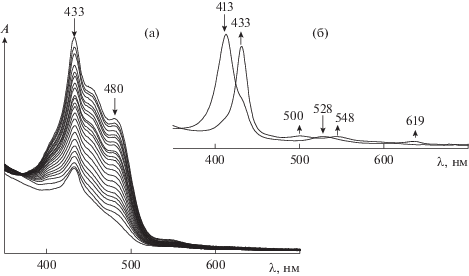
Рис. 2.
ИК-спектры (ацетонитрил) CoIITPP(OCH3)8 (1) и реакционной смеси CoIITPP(OCH3)8 с Н2О2 (2) (${{С}_{{{\text{C}}{{{\text{o}}}^{{{\text{II}}}}}{\text{TPP}}{{{\left( {{\text{OC}}{{{\text{H}}}_{{\text{3}}}}} \right)}}_{{\text{8}}}}}}}$ = 9.5 × 10–6 моль/л, ${{C}_{{{{{\text{H}}}_{{\text{2}}}}{{{\text{O}}}_{{\text{2}}}}}}}$ = 8 × × 10–1 моль/л).
В масс-спектре реакционной смеси (${{C}_{{{\text{CoTPP}}{{{\left( {{\text{OC}}{{{\text{H}}}_{{\text{3}}}}} \right)}}_{{\text{8}}}}}}}$ = 5.4 × 10–6 моль/л, ${{C}_{{{{{\text{H}}}_{{\text{2}}}}{{{\text{O}}}_{{\text{2}}}}}}}$ = 1.29 × × 10–1 моль/л) наблюдаются сигналы молекулярных пиков m/z = 913.9, 929.8, 944.8 и 960.8, соответствующие ионной форме комплексов (ОН)CoTPP(OCH3)7(О), (ОН)CoTPP(OCH3)7(ОО), (ОН)CoTPP(OCH3)7(ОСН2ОH) и (ОН)CoTPP(OCH3)7(ОСН2ООН) (рис. 3).
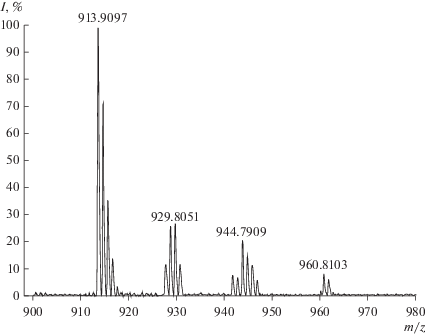
Полученные данные свидетельствуют о том, что в результате активации пероксида водорода образуется двуокисленная форма с радикалом на метокси группе $-{\text{ОСН}}_{2}^{\centerdot },$ которая под действием кислорода воздуха быстро трансформируется в радикал типа •ООR. Такие радикалы очень активны и способны окислять пероксид водорода до Н2О и О2, что и наблюдается в ходе реакции. Соединения, обнаруженные методом масс-спектрометрии (рис. 3), являются интермедиатами и продуктами этого редокс-процесса.
Образование двуокисленной формы порфирината кобальта(II) в рассматриваемых условиях происходит мгновенно, поэтому после регистрации ее ЭСП в раствор добавляли тетратерпен – каротин (Ccar = 2.3 × 10–4–5.9 × 10–4 моль/л) и ликопин (Cлик = 4.9 × 10–5–1.35 × 10–4 моль/л) и фиксировали изменения во времени оптической плотности в ЭСП смеси при 480 и 473 нм для каротина и ликопина соответственно (рис. 1). Степень деградации тетратерпенов в реакции окисления составляет 98% в течение 2–4 ч. В отсутствие порфирината кобальта(II) изменения в ЭСП смеси незначительные, деградация субстратов за 24 ч составляет лишь 5%.
Следует отметить, что окисление каротина идет параллельно с окислением пероксида водорода. Выделение О2 наблюдается в течение всей реакции. А вот при добавлении ликопина выделение кислорода прекращается через 1–2 мин.
С учетом спектральных характеристик окисление тетратерпенов пероксидом водорода в присутствии порфирината кобальта(II) описывается уравнением:
(1)
$\begin{gathered} {\text{C}}{{{\text{o}}}^{{{\text{II}}}}}{\text{TPP}}{{\left( {{\text{OC}}{{{\text{H}}}_{{\text{3}}}}} \right)}_{{\text{8}}}} + {\text{ }}{{{\text{H}}}_{{\text{2}}}}{{{\text{O}}}_{2}}\xrightarrow{{{{k}_{{{\text{v}}1}}}}} \\ \to {{\left[ {{\text{C}}{{{\text{o}}}^{{{\text{III}}}}}{\text{TPP}}{\kern 1pt} \left( {{\text{OC}}{{{\text{H}}}_{{\text{3}}}}} \right)_{8}^{ \bullet }} \right]}^{{2 + }}}\, + \,{\text{тетратерпен}}\xrightarrow{{{{k}_{{{\text{v}}2}}}}} \\ \to {\text{продукты}}\,\,{\text{окисления}}. \\ \end{gathered} $Эффективные константы скорости реакции формально первого порядка (lg kэф) в системе двуокисленная форма порфирината кобальта(II)–тетратерпен–ацетонитрил при постоянной концентрации комплекса коррелируют с lg Cтетратерп (рис. 4). Из линейной зависимости lg kэф от lg Cтетратерп по уравнению (2) определены порядки по субстратам (n = 0 для каротина и n = 1 для ликопина) с помощью константы скорости реакции (табл. 1):
Рис. 4.
Зависимость kэф–f(Cтетратерп) для реакции окисления тетратерпенов пероксидом водорода (${{C}_{{{{{\text{H}}}_{{\text{2}}}}{{{\text{O}}}_{{\text{2}}}}}}}$ = 1.4 моль/л) в присутствии CoIITPP(OCH3)8 (${{С}_{{{\text{C}}{{{\text{o}}}^{{{\text{II}}}}}{\text{TPP}}{{{\left( {{\text{OC}}{{{\text{H}}}_{{\text{3}}}}} \right)}}_{{\text{8}}}}}}}$ = 5.4 × 10–6 моль/л) без имидазола (1), с имидазолом (2) (CIm = 3.5 × × 10–3 моль/л) в ацетонитриле при 295 K: а – окисление каротина (Ccar = 1.67 × 10–4–5.87 × 10–4 моль/л), б – окисление ликопина (Cлик = 4.94 × 10–5–1.35 × 10–4 моль/л).
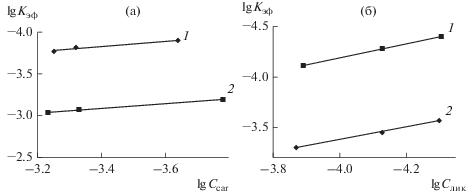
Таблица 1.
Кинетические параметры реакции окисления тетратерпенов пероксидом водорода в присутствии порфирината кобальта(II) в ацетонитриле при 295 K
| Сcar × 104, моль/л | kэф × 104, с–1 |
|---|---|
| ${{C}_{{{\text{C}}{{{\text{o}}}^{{{\text{II}}}}}{\text{TPP(OC}}{{{\text{H}}}_{{\text{3}}}}{{{\text{)}}}_{{\text{8}}}}}}}$ = 5.4 × 10–6 моль/л, ${{C}_{{{{{\text{H}}}_{2}}{{{\text{O}}}_{2}}}}}$ = 1.40 моль/л | |
| 2.31 | 1.26 |
| 4.80 | 1.53 |
| 5.63 | 1.70 |
| kv = 1.70 × 10–3 с–1 моль–1 л1 | |
| ${{C}_{{{\text{C}}{{{\text{o}}}^{{{\text{II}}}}}{\text{TPP(OC}}{{{\text{H}}}_{{\text{3}}}}{{{\text{)}}}_{{\text{8}}}}}}}$ = 5.4 × 10–6 моль/л, ${{C}_{{{{{\text{H}}}_{2}}{{{\text{O}}}_{2}}}}}$ = 1.40 моль/л, CIm = 3.5 × 10–3 моль/л | |
| 1.67 | 6.39 |
| 4.69 | 8.48 |
| 5.87 | 9.10 |
| kv = 7.20 × 10–3 с–1 моль–1 л1 | |
| Cлик × 105, моль/л | kэф × 105, с–1 |
| ${{C}_{{{\text{C}}{{{\text{o}}}^{{{\text{II}}}}}{\text{TPP(OC}}{{{\text{H}}}_{{\text{3}}}}{{{\text{)}}}_{{\text{8}}}}}}}$ = 5.4 × 10–6 моль/л, ${{C}_{{{{{\text{H}}}_{2}}{{{\text{O}}}_{2}}}}}$ = 1.40 моль/л | |
| 4.94 | 3.98 |
| 7.46 | 5.29 |
| 12.90 | 7.72 |
| kv = 3.70 × 10–2 с–1 моль–1 л1 | |
| ${{C}_{{{\text{C}}{{{\text{o}}}^{{{\text{II}}}}}{\text{TPP(OC}}{{{\text{H}}}_{{\text{3}}}}{{{\text{)}}}_{{\text{8}}}}}}}$ = 5.4 × 10–6 моль/л, ${{C}_{{{{{\text{H}}}_{2}}{{{\text{O}}}_{2}}}}}$ = 1.40 моль/л, CIm = 3.5 × 10–3 моль/л | |
| 4.94 | 27.40 |
| 7.46 | 36.39 |
| 13.50 | 51.00 |
| kv = 1.32 × 10–1 с–1 моль–1 л1 | |
| ${{C}_{{{\text{C}}{{{\text{o}}}^{{{\text{II}}}}}{\text{TPP}}}}}$ = 6.8 × 10–6 моль/л, ${{C}_{{{{{\text{H}}}_{2}}{{{\text{O}}}_{2}}}}}$ = 1.40 моль/л | |
| 4.51 | 1.54 |
| 6.43 | 1.63 |
| 11.20 | 1.94 |
| kv = 1.65 × 10–4 с–1 моль–1 л1 | |
Кинетические параметры, полученные в ходе исследований, позволяют записать скорость реакции окисления тетратерпенов в виде уравнений:
(4)
$ - d{{C}_{{{\text{лик}}}}}/d\tau = k{{C}_{{{\text{радикал}}{\text{.}}\,{\text{форма}}}}}{{С}_{{{\text{лик}}}}}.$Окислительное разложение каротиноидов представляет собой сложный и многостадийный процесс, в результате которого образуются такие интермедиаты, как моно- и бирадикальные формы, апокаротеналы, эпоксиды, аполикопины, аполикопиналы и др., ведущие к образованию множества низкомолекулярных продуктов [24–27, 30–32, 44]. Нулевой порядок по каротину можно объяснить участием в лимитирующей стадии реакции не исходного соединения, а продуктов его первичного окисления. В работе [27] при изучении реакций такого типа в среде органических растворителей под действием различных окислителей был также установлен нулевой порядок по β-каротину.
Анализ кинетических данных (табл. 1) показывает, что окисление ликопина идет в 20 раз быстрее, чем каротина. Данный факт хорошо согласуется с электрохимическими исследованиями окисления природных каротиноидов. В работе [45] показано, что первый и второй потенциалы окисления ликопина Е 0 = 0.50 и 0.52 В, а для каротина Е 0 = = 0.54 и 0.55 В соответственно.
Следует также учесть, что определенный вклад в скорость процесса окислительного разложения β-каротина вносят активные интермедиаты, образованные в результате окисления пероксида водорода активной формой порфирината кобальта(III) с радикалом на метоксигруппе (•OOR). Эта реакция идет параллельно, и интермедиаты могут принимать участие в окислении субстрата. Выступая инициаторами свободнорадикальных реакций, эти соединения сами становятся уязвимыми и подвергаются мономолекулярному или индуцированному распаду. Кроме того, активными интермедиатами реакции могут быть бирадикалы Крига [27], способные участвовать в дальнейшем окислении как в свободном виде, так и будучи координированными на катионе металла комплекса. Поэтому константа скорости окисления каротина является суммарной величиной. При разложении ликопина параллельной реакции окисления пероксида водорода не наблюдается. Причина этого в быстрой нейтрализации ликопином радикала •OOR, локализованного на периферии порфирината кобальта.
Окисление тетратерпенов, сопровождаемое частичным разрушением порфиринового хромофора комплекса, подобно ферментативному распаду гемма крови и деструкции хлорофилла [46, 47]. Оно связано с нарушением сопряженности в π-системе и разрывом одного из метиновых мостиков тетрапиррольного кольца по α- или мезо-положению за счет локализации в этом месте радикала или низкомолекулярного радикального фрагмента. В результате образуется нециклический тетрапиррол. Далее происходит его фрагментация до низкомолекулярных соединений [48, 49]. Степень деградации порфирината кобальта за время реакции окислительного разложения тетратерпенов составляет 10–15%. Конечный спектр реакционной смеси после окисления тетратерпенов соответствует двуокисленной форме порфирината кобальта ${{\left[ {{\text{C}}{{{\text{o}}}^{{{\text{III}}}}}{\text{TPP}}\left( {{\text{OC}}{{{\text{H}}}_{{\text{3}}}}} \right)_{8}^{ \bullet }} \right]}^{{2 + }}}.$ Восстановленную форму комплекса зафиксировать не удалось из-за высокой скорости образования радикальной формы.
Активность формы ${{\left[ {{\text{C}}{{{\text{o}}}^{{{\text{III}}}}}{\text{TPP}}\left( {{\text{OC}}{{{\text{H}}}_{{\text{3}}}}} \right)_{8}^{ \bullet }} \right]}^{{2 + }}}$ подтверждается дальнейшим протеканием реакции разложения тетратерпенов без добавок пероксида водорода. При последующем добавлении в конечный раствор очередной порции субстрата появившиеся вновь полосы β-каротина и ликопина при 480 и 473 нм снова исчезают с сохранением полос окисленной формы комплекса, интенсивность которых существенно уменьшается после проведения 5 циклов. Константа скорости окислительного разложения тетратерпенов при этом практически не изменяется, о чем свидетельствуют кинетические кривые процесса (рис. 5).
Рис. 5.
Кинетические кривые реакции окислительного разложения ликопина (Cлик = 7.46 × 10–5 моль/л) пероксидом водорода (${{C}_{{{{{\text{H}}}_{{\text{2}}}}{{{\text{O}}}_{{\text{2}}}}}}}$ = 1.4 моль/л) в присутствии CoIITPP(OCH3)8 (${{С}_{{{\text{C}}{{{\text{o}}}^{{{\text{II}}}}}{\text{TPP}}{{{\left( {{\text{OC}}{{{\text{H}}}_{{\text{3}}}}} \right)}}_{{\text{8}}}}}}}$ = 5.4 × 10–6 моль/л) при периодическом добавлении субстрата в реакционную смесь: а – без добавления имидазола, б – с добавлением имидазола (CIm = 3.5 × × 10–3 моль/л).
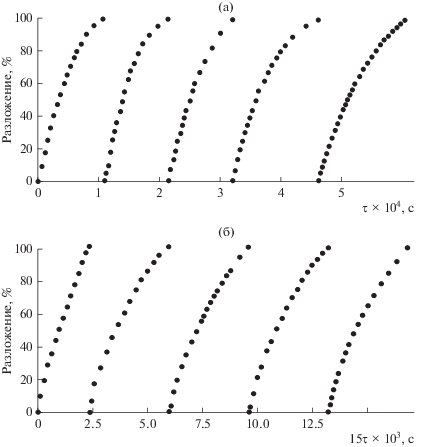
Как известно, находящиеся в транс-положении к активному центру аксиальные лиганды на катионе металла определяют активность макроциклических тетрапиррольных соединений при связывании и транспортировке газов, переносе электронов и катализе [4, 5, 8, 50–52]. Поэтому было исследовано влияние присутствия имидазола на скорость окислительной деструкции тетратерпенов. Введение имидазола (Im, CIm = 3.5 × 10–3 моль/л) в состав координационной сферы соединения сопровождается образованием донорно-акцепторного комплекса [53, 54]. Добавление пероксида водорода ведет к его окислению и образованию активной формы (ЭСП, λmax = 433, 546 и 616 нм). Скорость реакции окислительного разложения тетратерпенов с участием этой частицы увеличивается в несколько раз (табл. 1). Однако донорно-акцепторный комплекс при активации пероксида водорода образует форму, которая не окисляет Н2О2 до Н2О и О2. Устойчивость активной частицы подтверждается дальнейшим протеканиием реакции окисления тетратерпенов без дополнительных добавок пероксида водорода с практически равными скоростями (рис. 5). Следует отметить, что введение имидазола стабилизирует комплекс, и его деградация составляет не более 2% при проведении 5 циклов окисления субстратов. Природа активной формы лигандированного порфирината кобальта является предметом отдельного исследования, которое будет проведено нами в дальнейшем.
Для установления зависимости каталитической активности от наличия дополнительных редокс-активаторов, а именно метоксильных заместителей, нами изучена реакция окислительного разложения ликопина (Cлик = 4.5 × 10–5–1.12 × 10–4 моль/л) пероксидом водорода (${{C}_{{{{{\text{Н}}}_{{\text{2}}}}{{{\text{О}}}_{{\text{2}}}}}}}$ = 1.4 моль/л) в присутствии незамещенного тетрафенилпорфирината кобальта(II) (CoTPP, CCoTPP = 6.8 × 10–6 моль/л). Окисленный комплекс [СоТРР•]2+ имеет ЭСП (λmax = 431, 498, 546, 616 нм), сходный со спектром ${{\left[ {{\text{C}}{{{\text{o}}}^{{{\text{III}}}}}{\text{TPP(OC}}{{{\text{H}}}_{{\text{3}}}}{\text{)}}_{8}^{ \bullet }} \right]}^{{2 + }}}.$ Это свидетельствует об одинаковом электронном состоянии молекул. Однако электрохимическое исследование показывает, что второй потенциал, при котором идет окисление порфиринового лиганда L ↔ L+ для СоТРР (Е1/2 = 1.19 В, радикал локализуется на порфириновом кольце [55]) значительно выше, чем для CoTPP(OCH3)8 (Е1/2 = 0.62 В, радикал локализуется на CH3O-заместителе). Эти данные позволяют говорить о разной локализации радикала в макроцикле. Комплекс [СоТРР•]2+ не является такой активной активной частицей [56], как ${{\left[ {{\text{C}}{{{\text{o}}}^{{{\text{III}}}}}{\text{TPP(OC}}{{{\text{H}}}_{{\text{3}}}}{\text{)}}_{8}^{ \bullet }} \right]}^{{2 + }}},$ и не окисляет пероксид водорода до Н2О и О2. Скорость реакции окисления ликопина с участием СоТРР почти на два порядка ниже, чем в присутствии CoTPP(OCH3)8 (табл. 1). Порядок по ликопину в этом случае равен 0.
ЗАКЛЮЧЕНИЕ
Наличие метоксигрупп, выступающих в качестве редокс-активаторов, и изменение состава координационного центра путем введения дополнительных лигандов оказывают существенное влияние на каталитическую активность порфирината кобальта(II) в реакции разложения тетратерпенов. Высокая реакционная способность двуокисленной радикальной формы порфирината кобальта в отношении пероксида водорода и активной формы лигандированного комплекса кобальта в окислительной деструкции тетратерпенов позволяет рассматривать эти соединения в качестве перспективных катализаторов окислительных процессов и моделей природных ферментов.
Список литературы
Patel M., Day B.J. // Trends Pharmacol. Sci. 1999. V. 20. № 9. P. 359. https://doi.org/10.1016/S0165-6147(99)01336-X
Dismukes G.C. // Chem. Rev. 1996. V. 96. № 7. P. 2909. https://doi.org/10.1021/cr950053c
Avdeev M.V., Bagri, E.I., Maravin G.B. et al. // Kinet. Catal. 2002. V. 43. № 1. P. 38. [Авдеев М.В., Багрий Е.И., Маравин Г.В. и др. // Кинетика и катализ. 2002. Т. 43. № 1. С. 43.]https://doi.org/10.1023/A:1014240927361
Capobianchi A., Paoletti A.M., Rossia G. et al. // Sens. Actuators, B. 2009. V. 142. № 1. P. 159. https://doi.org/10.1016/j.snb.2009.08.021
Zanotti G., Angelini N., Notarantonio S. et al. // Int. J. Photoenergy. 2010. V. 2010. Article ID 136807. https://doi.org/10.1155/2010/136807
Yusubov M.S., Celik C., Geraskina M.R. et al. // Tetrahedron Lett. 2014. V. 55. № 41. P. 5687. https://doi.org/10.1016/j.tetlet.2014.08.070
Chapman C.M., Pruneau J.M., Laverack C.A. et al. // App. Catal., A. 2016. V. 510. P. 204. https://doi.org/10.1016/j.apcata.2015.11.031
Oszajca M., Franke A., Brindell M. et al. // Coord. Chem. Rev. 2016. V. 306. Part 2. P. 483. https://doi.org/10.1016/j.ccr.2015.01.013
Quesne M.G., Senthilnathan D., Singh D. et al. // ACS Catal. 2016. V. 6. № 4. P. 2230. https://doi.org/10.1021/acscatal.5b02720
Zaitseva S.V., Zdanovich S.A., Kudrik E.V. et al. // Russ. J. Inorg. Chem. 2017. V. 62. № 9. P. 1257. [Зайцева С.В., Зданович С.А., Кудрик Е.В. и др. // Журн. неорган. химии. 2017. Т. 62. № 9. С. 1265.]https://doi.org/10.1134/S0036023617090194
Tyulyaeva E.Yu. // Russ. J. Inorg. Chem. 2019. V. 64. № 14. P. 1775. https://doi.org/10.1134/S0036023619140110
Bharati S.L., Sarma C., Hazarika P.J. et al. // Russ. J. Inorg. Chem. 2019. V. 64. № 3. P. 335. https://doi.org/10.1134/S0036023619030045
Bajju G.D., Devi G., Ahmed A. et al. // Russ. J. Inorg. Chem. 2019. V. 64. № 6. P. 734. https://doi.org/10.1134/S0036023619060044
Song E., Shi C., Anson F.C. // Langmuir. 1998. V. 14. № 15. P. 4315. https://doi.org/10.1021/la980084d
Shi C., Anson F.C. // Inorg. Chem. 1998. V. 37. № 5. P. 1037. https://doi.org/10.1021/ic971255p
Милаева Е.Р. // Рос. хим. журн. 2004. Т. 48. № 4. С. 20.
Troja’nek A., Langmaier J., Samec Z. // Electrochem. Commun. 2006. V. 8. № 3. P. 475. https://doi.org/10.1016/j.elecom.2006.01.004
Antonova N.A., Osipova V.P., Kolyada M.N. et al. // Macroheterocycles. 2010. V. 3. № 2-3. P. 139. [Антонова Н.А., Осипова В.П., Коляда М.Н. и др. // Макрогетероциклы. 2010. Т. 3. № 2–3. С. 139.]https://doi.org/10.6060/mhc2010.2-3.139
Zaitseva S.V., Simonova O.R., Zdanovich S.A., Koifman O.I. // Macroheterocycles. 2018. V. 11. № 1. P. 29. [Симонова О.Р., Зайцева С.В., Зданович С.А., Койфман О.И. // Макрогетероциклы. 2018. V. 11. № 1. P. 29.]https://doi.org/10.6060/mhc180173s
Grishina E.S., Kudrik E.V., Makarova A.S. et al. // Russ. J. Phys. Chem. A. 2016. V. 90. № 3. P. 704. [Гришина Е.Г., Макарова А.С., Кудрик Е.В. и др. // Журн. физ. химии. 2016. Т. 90. № 3. С. 477.]https://doi.org/10.1134/S0036024416030134
Simonova O.R., Zaitseva S.V., Tyulyaeva E.Yu. et al. // Russ. J. Inorg. Chem. 2017. V. 62. № 4. P. 508. [Симонова О.Р., Зайцева С.В., Тюляева Е.Ю. и др. // Журн. неорган. химии. 2017. Т. 62. № 4. С. 509.]https://doi.org/10.1134/S0036023617040179
Zaitseva S.V., Tyulyaeva E.Yu., Simonova O.R. et al. // J. Coord. Chem. 2018. V. 71. № 16–18. P. 2995. https://doi.org/10.1080/00958972.2018.1506109
Zaitseva S.V., Tyulyaeva E.Yu., Zdanovich S.A., Koifman O.I. // J. Mol. Liq. 2019. V. 287. P. 111023. https://doi.org/10.1016/j.molliq.2019.111023
Malakhova M.V., Orlova V.F., Karpov V.A. et al. // Bull. Exp. Bio. Med. Biophys. Biochem. 1998. V. 126. № 3. P. 928. https://doi.org/10.1007/BF02447377
Caris-Veyrat C., Amiot M.-J., Ramasseul R., Marchon J.-C. // New J. Chem. 2001. V. 25. № 2. P. 203. https://doi.org/10.1039/B008378J
Kodis G., Liddell P.A., Moore A.L. et al. // J. Phys. Org. Chem. 2004. V. 17. № 9. P. 724. https://doi.org/10.1002/poc.787
de Jesus Benevides C.M., da Cunha Veloso M.C., de Paula Pereira P.A., de Andrade J.B. // Food Chem. 2011. V. 126. № 3. P. 927. https://doi.org/10.1016/j.foodchem.2010.11.082
Adler A.D., Longo F.R., Kampas F., Kim J. // J. Inorg. Nucl. Chem. 1970. V. 32. № 7. P. 2443. https://doi.org/10.1016/0022-1902(70)80535-8
Экспериментальные методы химической кинетики / Под ред. Эмануэля Н.М., Сергеева Г.Б. М.: Высш. шк., 1980. 375 с.
Kennedy T.A., Liebler D.C. // Chem. Res. Toxicol. 1991. V. 4. № 3. P. 290. https://doi.org/10.1021/tx00021a005
Yamauchi R., Miyake N., Inoue H., Kato K. J. // Agric. Food Chem. 1993. V. 41. № 5. P. 708. https://doi.org/10.1021/jf00029a005
French R.R., Holzer P., Leuenberger M.G., Woggon W.-D. // Angew. Chem. Int. Ed. 2000. V. 39. № 7. P. 1267. https://doi.org/10.1002/(SICI)1521-3773(20000403)39:7< 1267::AID-ANIE1267>3.0.CO;2-7
Isci U., Dumoulin F., Sorokin A.B., Ahsen V. // Turk. J. Chem. 2014. V. 38. P. 923. https://doi.org/10.3906/kim-1407-47
Sorokin A.B., Kudrik E.V. // Catalysis Today. 2011. V. 159. № 1. P. 37. https://doi.org/10.1016/j.cattod.2010.06.020
Makarova A.S., Kudrik E.V., Makarov S.V., Koifman O.I. // J. Porphyrins Phthalocyanines. 2014. V. 18. № 7. P. 604. https://doi.org/10.1142/S1088424614500369
Afanasiev P., Sorokin A.B. // Acc. Chem. Res. 2016. V. 49. № 4. P. 583. https://doi.org/10.1021/acs.accounts.5b00458
Colomban C., Kudrik E.V., Tyurin D. V. et al. // Dalton Trans. 2015. V. 44. № 5. P. 2240. https://doi.org/10.1039/c4dt03207a
Patil A.O., Curtin D.Y., Paul I.C. // J. Am. Chem. Soc. 1984. V. 106. № 2. P. 348. https://doi.org/10.1021/ja00314a017
Yoshida R., Isozaki K., Yokoi T. et al. // Org. Biomol. Chem. 2016. V. 14. № 31. P. 7468. https://doi.org/10.1039/c6ob00969g
Oxley J., Smith J., Brady J. et al. // Appl. Spectrosc. 2008. V. 62. № 8. P. 906. https://doi.org/10.1366/000370208785284420
Pettersson M., Tuominen S., Räsänen M. // J. Phys. Chem. A. 1997. V. 101. № 6. P. 1166. https://doi.org/10.1021/jp962946u
Amanullah S., Singha A., Dey A. // Coord. Chem. Rev. 2019. V. 386. P. 183. https://doi.org/10.1016/j.ccr.2019.01.021
Kitagawa T., Ozaki Y. // Metal Complexes with Tetrapyrrole Ligands I. / Ed. Buchler J.W. Berlin: Springer, 1987. P. 71. https://doi.org/10.1007/BFb0036790
Carail M., Caris-Veyrat C. // Pure Appl. Chem. 2006. V. 78. № 7. P. 1493. https://doi.org/10.1351/pac200678071413
Liu D., Gao Y., Kispert L.D. // J. Electroanal. Chem. 2000. V. 488. № 2. P. 140. https://doi.org/10.1016/S0022-0728(00)00205-9
Sugishima M., Sakamoto H., Higashimoto Y. et al. // J. Biol. Chem. 2003. V. 278. № 34. P. 32352. https://doi.org/10.1074/jbc.M303682200
Zhang H., Liu N., Zhao J. et al. // Chemosphere. 2019. V. 223. P. 659. https://doi.org/10.1016/j.chemosphere.2019.01.135
Müller T., Rafelsberger M., Vergeiner C., Kräutler B. // Angew. Chem. Int. Ed. 2011. V. 50. № 45. P. 10724. https://doi.org/10.1002/anie.201103934
Simonova O.R., Zaitseva S.V., Koifman O.I. // Russ. J. Gen. Chem. 2016. V. 86. № 6. С. 1322. [Симонова О.Р., Зайцева С.В., Койфман О.И. // Журн. общ. химии. 2016. Т. 86. № 6. С. 992.]https://doi.org/10.1134/S1070363216060177
Zaitseva S.V., Zdanovich S.A., Tyulyaeva E.Yu. et al. // J. Porphyrins Phthalocyanines. 2016. V. 20. № 5. P. 639. https://doi.org/10.1142/S1088424616500474
Zaitseva S.V., Zdanovich S.A., Kudrik E.V., Koifman O.I. // Russ. J. Inorg. Chem. 2017. V. 62. № 9. P. 1257 [Зайцева С.В., Зданович С.А., Кудрик Е.В., Койфман О.И. // Журн. неорган. химии. 2017. Т. 62. № 9. С. 1265.]https://doi.org/10.1134/S0036023617090194
Motorina E.V., Mozhzhukhina E.G., Lomova T.N. // J. Struct. Chem. 2018. V. 59. № 8. P. 1880. [Моторина Е.В., Можжухина Е.Г., Ломова Т.Н. // Журн. структур. химии. 2018. Т. 59. № 8. С. 1942.]https://doi.org/10.1134/S0022476618080164
Zakavi S., Talebzadeh S., Rayati S. // Polyhedron. 2012. V. 31. № 1. P. 368. https://doi.org/10.1016/j.poly.2011.09.038
Zaitseva S.V., Zdanovich S.A., Koifman O.I. // Macroheterocycles. 2012. Т. 5. № 1. С. 81. [Зайцева С.В., Зданович С.А., Койфман О.И. // Макрогетероциклы. 2012. Т. 5. № 1. С. 81.]https://doi.org/10.6060/mhc2012.111149z
Wolberg A., Manassen J. // J. Am. Chem. Soc. 1970. V. 92. № 10. P. 2982. https://doi.org/10.1021/ja00713a010
Wang D., Groves J.T. // PNAS. 2013. V. 110. № 39. P. 15 579. https://doi.org/10.1073/pnas.1315383110
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии