

Журнал неорганической химии, 2020, T. 65, № 6, стр. 770-777
Синтез новых борсодержащих лигандов и комплексов гафния(IV) на их основе
В. В. Воинова a, И. Н. Клюкин a, А. П. Жданов a, *, М. С. Григорьев b, К. Ю. Жижин a, Н. Т. Кузнецов a
a Институт общей и неорганической химии им. Н.С. Курнакова РАН
119991 Москва, Ленинский пр-т, 31, Россия
b Институт физической химии и электрохимии им. А.Н. Фрумкина РАН
119991 Москва, Ленинский пр-т, 31, Россия
* E-mail: zhdanov@igic.ras.ru
Поступила в редакцию 02.12.2019
После доработки 27.01.2020
Принята к публикации 30.01.2020
Аннотация
Предложен двустадийный способ синтеза борсодержащего комплексона на основе аминомалоновой кислоты. Продукты на всех стадиях синтеза исследованы методами мультиядерной спектроскопии ЯМР, ИК-спектроскопии поглощения, ESI масс-спектрометрии. Для продукта (NBu4)[2-B10H9NHC(CH3)NHCH(COOEt)2] структура подтверждена методом РСА монокристалла. Изучена реакция комплексообразования борилированного производного аминомалоновой кислоты с Hf(NEt2)4 и Hf(OBu)4.
ВВЕДЕНИЕ
Кластерные анионы бора и соединения на их основе широко применяются в качестве прекурсоров для получения боридов [1], магнитных устройств [2, 3] и элементов устройств фотовольтаики [4] и оптики [5, 6]. Соединения, содержащие клозо-декаборатный анион, используются для создания новых лекарственных препаратов для 10B-нейтронозахватной терапии (БНЗТ), которая успешно применяется для лечения неоперабельных злокачественных опухолей [7, 8]. Посредством БНЗТ осуществляется таргетное химиотерапевтическое воздействие на опухолевые клетки [9–11]. Эффективность борсодержащего препарата зависит от ряда важных свойств: низкой токсичности, возможности получения водорастворимых форм, а также от высокой степени селективности накопления в поврежденных клетках [9]. Поэтому основное внимание в ходе исследований было направлено на разработку методов функционализации кластерных анионов бора. Наибольшее распространение получили следующие методы: раскрытие циклических ониевых производных [12–15], прямая функционализация [16, 17], ипсо-замещение [18–22], присоединение к кратным связям (нуклеофильное присоединение к экзополиэдрическому нитрилиевому заместителю) [23–32].
Наряду с БНЗТ активно развиваются и другие способы лечения неоперабельных злокачественных опухолей. Так, в фотонзахватной терапии (ФЗТ) опухолей использование наночастиц с высоким зарядом ядра, так называемых радиосенсибилизаторов, повышающих чувствительность клеток к радиации, позволяет повысить терапевтическую эффективность за счет избирательного усиления эффектов радиационного повреждения в опухоли по сравнению со здоровыми тканями. На сегодняшний день большинство исследований направлено на изучение радиовоздействия с помощью наночастиц золота [33–35], гадолиния [36], платины, оксида железа и гафния. Выявлено значительное увеличение ингибирования роста опухоли у мышей с предварительно ксенотрансплантированными опухолями, получавших лучевую терапию и препарат, содержащий наночастицы золота [37–40] или гафния [41, 42], по сравнению с особями, подвергшимися лишь лучевой терапии.
В связи с этим в настоящей работе нами предложены способы получения соединений, которые могут использоваться для комбинированной терапии ФЗТ-БНЗТ благодаря содержанию в них как атомов бора, так и иона гафния(IV).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Элементный анализ на углерод, водород и азот осуществляли на автоматическом газовом анализаторе CHNS-3 FA 1108 Elemental Analyser (Carlo Erba). Определение бора и гафния методом ICP MS выполнено на атомно-эмиссионном спектрометре с индуктивно-связанной плазмой iCAP 6300 Duo в ЦКП “Научно-аналитического центра ФГУП “ИРЕА” Национального исследовательского центра Курчатовский институт”.
ИК-спектры соединений записывали на ИК-Фурье-спектрофотометре Инфралюм ФТ08 (НПФ АП “Люмекс”) в области 4000–400 см–1 с разрешением 1 см–1. Образцы готовили в виде раствора в хлороформе.
Спектры ЯМР 1H, 11B, 13C растворов исследуемых веществ в CD3CN записывали на импульсном Фурье-спектрометре Bruker MSL-300 (Германия) на частотах 300.3, 96.32 и 75.49 МГц соответственно с внутренней стабилизацией по дейтерию. В качестве внешних стандартов использовали тетраметилсилан или эфират трехфтористого бора.
Масс-спектры растворов исследуемых веществ в CH3CN записывали на спектрометре API 3200 Qtrap (AppliedBiosysteм, USA). Условия ионизации: турбоионное распыление, ионное распыление, напряжение ±4500 В, декластеризации ±12 В, скорость потока 2–20 мкл/мин. Средняя аналитическая концентрация образцов 0.5–1.0 мг/л.
Рентгеноструктурный анализ соединения 2 выполнен в ЦКП ИФХЭ РАН на автоматическом четырехкружном дифрактометре с двумерным детектором Bruker KAPPA APEX II (излучение MoKα) [43] с использованием фрагмента кристалла размерами 0.12 × 0.08 × 0.04 мм при температуре 100 K.
Параметры элементарной ячейки уточнены по всему массиву данных [44]. Структура расшифрована прямым методом [45] и уточнена полноматричным методом наименьших квадратов [46] по F2 по всем данным в анизотропном приближении для всех неводородных атомов (кроме разупорядоченных, если такие имеются). Атомы H кластера бора локализованы из разностного Фурье-синтеза электронной плотности и уточнены изотропно без каких-либо ограничений. Атомы H групп NH, CH, CH2 и CH3 размещены в геометрически вычисленных позициях и уточнены с изотропными температурными параметрами, равными 1.2Uэкв атома N или C для NH, CH, CH2 и 1.5Uэкв атома C для CH3.
Растворители и реагенты марки “х. ч.” и “ос. ч.” использовали без дополнительной очистки.
(NBu4)[2-B10H9(NCCH3)] (1) получали по известной методике [47]. Растворяли 10.00 г (0.017 моль) (NBu4)2[B10H10] в 50 мл CH3CN и добавляли 5 мл CF3COOH. Раствор нагревали до 60°C в атмосфере сухого аргона при перемешивании в течение 2 ч до прекращения газовыделения. Охлажденный до комнатной температуры раствор концентрировали на роторном испарителе. Сконцентрированный раствор разбавляли 20 мл ледяной уксусной кислоты и фильтровали через фильтр Шотта. Осадок на фильтре промывали 50 мл ледяной уксусной кислоты и 50 мл диэтилового эфира. Осадок высушивали в эксикаторе над P2O5. Получено 6.58 г (0.019 моль) (NBu4)[2-B10H9(NCCH3)] (98.9%).
(NBu4)[2-B10H9NHC(CH3)NHCH(COOEt)2] (2). Навеску 0.250 г (2 ммоль) NH2CH(COOEt)2 растворяли в 20 мл CH2Cl2 и добавляли производное 1 (0.400 г, 1 ммоль). Реакционную массу кипятили в течение 4 ч в колбе, снабженной магнитной мешалкой и обратным холодильником. После охлаждения до комнатной температуры реакционную массу экстрагировали 15 мл 0.1 н раствора соляной кислоты, а затем трижды промывали дистиллированной водой. Органический слой отделяли, сушили над безводным Na2SO4 и упаривали на роторном испарителе. Получено 0.539 г (NBu4)[2-B10H9NHC(CH3)NHCH(COOEt)2] (93.9%).
ИК-спектр (CHCl3), см–1: 3391 (ν(N–H)), 2470 (ν(B–H)), 1700 (ν(C=O)), 1645 (ν(С=N)), 1265 (ν(CO–O)). 1H ЯМР (δ, м.д.): 8.93 (уш. с, 1H, ‒NH–C(CH3)–NH–CH–), 6.67 (уш. с, 1H, ‒NH–CCH3), 4.96 (с, 1H, –CH–COOEt), 4.32 (кв, 2H, –COO–CH2–CH3, J = 8 Гц), 3.08 (8H, ${\text{NBu}}_{{\text{4}}}^{{\text{ + }}}$), 1.99 (с, 3H, –NH–C–CH3), 1.60 (8H, ${\text{NBu}}_{{\text{4}}}^{{\text{ + }}}$), 1.31 (т, 6H, –COO–CH2–CH3, 8 Гц), 0.96 (12H, ${\text{NBu}}_{{\text{4}}}^{{\text{ + }}}$). 11B{1H} ЯМР (δ, м.д.): 1.1 (с, 1B, B(10)), –5.9 (с, 1B, B(1)), –16.9 (с, 1B, B(2)), –25.6 (4B, B(3, 5–7), ‒28.7 (3B, B(4, 8–9). 13C ЯМР (δ, м.д.): 166.26 (‒COOEt), 165.91 (–NH–C–CH3), 63.87 (‒COOCH2CH3), 60.68 (–NH–CH–COOEt), 19.48 (–NH–C–CH3), 14.30 (–COOCH2CH3). MS (ESI) m/z = 333.2 а.е.м (соответствует пику молекулярного иона [B10H9NHC(CH3)NHCH(COOEt)2]–, для {[A]–} вычислено 333.3).
| C | H | N | B | |
| Найдено, %: | 51.88; | 10.61; | 7.22; | 19.2. |
| Для C25H61B10N3O4 (М = 575.9) | ||||
| вычислено, %: | 51.94; | 10.64; | 7.27; | 19.1. |
(NBu4)[2-B10H9NHC(CH3)NHCH(COOH)2] (3). Растворяли 0.500 г соединения 2 в 30 мл смеси CH3CN и концентрированной соляной кислоты в соотношении 4 : 1. Реакционную массу кипятили в течение 2 ч в колбе, снабженной магнитной мешалкой и обратным холодильником. После охлаждения до комнатной температуры реакционную массу концентрировали на роторном испарителе и трижды экстрагировали 15 мл CH2Cl2. Объединенные экстракты сушили над безводным Na2SO4 и упаривали на роторном испарителе. Получено 0.413 г (Bu4N)[2-B10H9NHC(CH3)NHCH(COOH)2] (89.3%).
ИК-спектр (CHCl3), см–1: 3491 (ν(O–H)), 3402 (ν(N–H)), 2464 (ν(B–H)), 1730 (ν(C=O)), 1659 (ν(С=N)), 1266 (ν(CO–O)). 1H ЯМР (δ, м.д.): 7.80 (уш. с, 1H, –NH–C(CH3)–NH–CH–), 6.88 (уш. с, 2H, –COOH), 6.19 (уш. с, 1H, –NH–C–CH3), 5.42 (с, 1H, NH–CH–COOH), 3.08 (8H, ${\text{NBu}}_{{\text{4}}}^{{\text{ + }}}$), 1.91 (c, 3H, NCCH3), 1.58 (8H, NBu4+), 1.34 (8H, ${\text{NBu}}_{{\text{4}}}^{{\text{ + }}}$), 0.93 (12H, ${\text{NBu}}_{{\text{4}}}^{{\text{ + }}}$). 11B{1H} ЯМР (δ, м.д.): 1.3 (с, 1B, B(10)), –5.2 (с, 1B, B(1)), –16.0 (с, 1B, B(2)), –25.0 (4B, B(3, 5–7), –28.2 (3B, B(4, 8–9). 13C ЯМР (δ, м.д.): 165.82 (–NH–C–CH3, –COOH), 54.45 (–NH–CH–COOH), 19.84 (–NH–C–CH3). MS (ESI) m/z = 279.2 (соответствует пику молекулярного иона [B10H9NHC(CH3)NHCH(COOH)2]–, для {[A]–} вычислено 279.2).
| C | H | N | B | |
| Найдено, %: | 48.21; | 10.21; | 8.01; | 21.0. |
| Для C21H53B10N3O4 (М = 521.5) | ||||
| вычислено, %: | 48.32; | 10.24; | 8.06; | 21.1. |
(NBu4)2[{2-B10H9NHC(CH3)NHCH(COO)2}2Hf] (4). Растворяли 0.200 г (0.4 ммоль) соединения 3 в 25 мл ТГФ и добавляли 0.180 г Hf(NEt2)4/0.236 г Hf(OBu)4 (0.5 ммоль). Реакционную массу перемешивали при комнатной температуре в атмосфере сухого аргона в течение 20 мин в колбе, снабженной магнитной мешалкой и обратным холодильником. Затем реакционную массу концентрировали на роторном испарителе. Получено 0.341 г (Bu4N)2[{B10H9NHC(CH3)NHCH(COO)2}2Hf] (91.0%).
ИК-спектр (CHCl3), см–1: 3491 (ν(O–H)), 3370 (ν(N–H)), 2466 (ν(B–H)), 1700 (ν(C=O)), 1663 (ν(С=N)), 1267 (ν(CO–O)). MS (ESI) m/z = 594.7 а. е. м. (соответствует пику иона {Hf(B10H9NHCCH3NHCH(COO)2)CH2(COO)2(H2O)2}–, вычислено 595.1), 228.2 а. е. м. (соответствует пику иона {Hf(B10H9NHCCH3NHCH(COO)2)}2–, вычислено 228.6).
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
В работе нами предложен метод синтеза нового борсодержащего комплексона на основе процессов нуклеофильного присоединения к кратным связям нитрилиевых производных клозо-декаборатного аниона (схема 1 ).
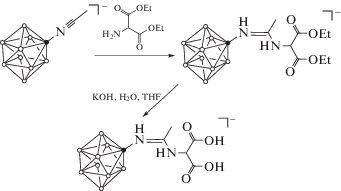
Схема 1 . Общая схема получения производного аминомалоновой кислоты.
На первой стадии процесса проводили нуклеофильное присоединение диэтиламиномалоната к производному клозо-декаборатного аниона на основе ацетонитрила, на второй стадии – щелочной гидролиз сложноэфирных групп. За ходом процесса следили с помощью спектроскопии 11B ЯМР. В спектрах продукта присоединения диэтиламиномалоната сигналы от апикальных атомов бора находятся в области 1.1 м.д. [B(10), I = 1] и –5.9 м.д. [B(1), I = 1], что характерно для продуктов такого типа [24]. Сигнал от замещенного атома бора B(2) наблюдается при –16.9 м.д., сигналы от незамещенных экваториальных вершин борного кластера – при –25.6 и –28.7 м.д. Процесс омыления сложноэфирных групп слабо сказывается на общем виде 11B ЯМР-спектра продукта на основе аминомалоновой кислоты, поэтому ход процесса контролировали с помощью тонкослойной хроматографии.
Строение полученных продуктов присоединения диэтиламиномалоната к нитрилиевому производному клозо-декаборатного аниона и продукта его гидролиза устанавливали с помощью мультиядерной спектроскопии ЯМР. Так, в спектре 1Н ЯМР соединения 2 амидиновый фрагмент представлен двумя сигналами: от протона иминогруппы при 6.67 м. д. и протона аминогруппы при 8.93 м. д. Диэтилмалонатный фрагмент представлен сигналами от метинового протона при 4.96 м. д., протонов этильных групп при 4.32 м.д. (квартет) и 1.31 м. д. (триплет). Сигналы протонов от заместителя нитрилиевой группы проявляются при 1.99 м. д. В спектрах присутствуют также сигналы от н-тетрабутиламмониевого катиона. В спектре 13С ЯМР диэтилмалонатный остаток представлен сигналами от атома углерода карбоксильной группы при 166.3 м. д., α-атома углерода при 60.7 м.д. и углеродных атомов этильных групп при 63.9 и 14.3 м. д. Для функциональной группы на основе ацетонитрила наблюдается два типа сигналов: при 165.9 м. д. для четвертичного атома углерода и при 19.5 м. д. для метильной группы.
По сравнению с соединением 2 в спектрах ЯМР продукта гидролиза наблюдаются некоторые изменения. Так, в спектрах ЯМР 1H исчезают сигналы от протонов этильной группы и появляется уширенный синглет от протонов карбоксильной группы при 6.88 м. д. В спектрах 13С сигнал от карбоксильного атома сливается с сигналом от четвертичного атома углерода нитрилиевой функциональной группы при 165.82 м. д.
Структура соединения 2 была подтверждена методом РСА монокристалла. Рис. 1 и 2 получены с помощью программного пакета OLEX2 [48]. Анионные фрагменты структуры представлены амидин-клозо-декаборатами. Заместитель находится в экваториальной позиции и имеет Z-конфигурацию двойной связи N(1)=C(1). Длина связи бор–азот (1.530 Å) указывает на ее ординарный характер. Фрагмент N(1)C(1)N(2)C(3) плоский (стандартное отклонение атомов от плоскости составляет 1.8°), а связи C(1)N(1) и C(1)N(2) существенно укорочены (1.297 и 1.329 Å соответственно), что указывает на наличие сопряжения и частичной делокализации положительного заряда на атомах N(1)C(1)N(2). Атом C(1) находится в sp2-гибридизации, углы N(1)C(1)N(2), N(1)C(1)C(2) и N(2)C(1)C(2) составляют 119.6(3)°, 120.7(3)° и 119.7(3)° соответственно. Структурные параметры остатка малонового эфира согласуются с данными для сложноэфирных групп [49].
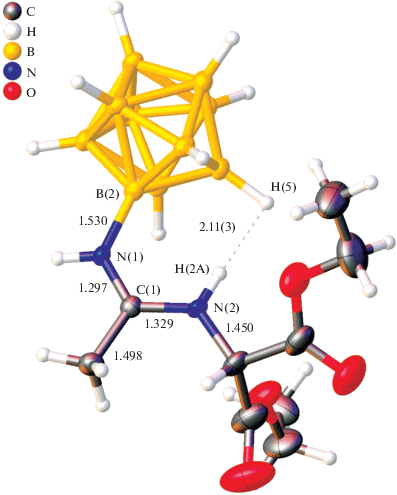
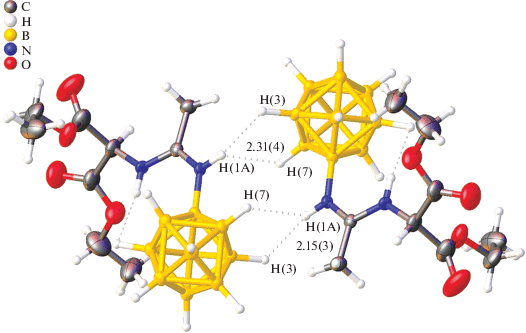
В структуре амидина заместитель стабилизирован диводородным взаимодействием между атомом водорода аминогруппы N(2)H(2) и гидридным атомом водорода H(5)B(5). Длина контакта составляет 2.11 Å. Межмолекулярные диводородные связи объединяют анионы в центросимметричные димеры (рис. 2).
Для полученного борсодержащего комплексона изучены процессы комплексообразования с участием Hf4+. В качестве гафниевых прекурсоров использовали бутоксид гафния(IV) и диэтиламид гафния(IV), что позволило провести реакции в среде эфирных растворителей и избежать процессов гелеобразования. Процесс комплексообразования контролировали по данным ИК-спектроскопии. Так, в ИК-спектрах поглощения полученных комплексов исчезает полоса поглощения валентных колебаний связи OH в области 3491 см–1, наблюдается также смещение полосы поглощения валентных колебаний связи C=O в область меньших волновых чисел – из области 1730 см–1 для исходного комплексона в область 1700 см–1 для полученных комплексных соединений. Образование комплексов подтверждено также методом ESI масс-спектрометрии. Так, в масс-спектрах продуктов присутствуют пики ионов {Hf(B10H9NHCCH3NHCH(COO)2)}2– при 228.2 а.е.м. (вычислено 228.6) и {Hf(B10H9NHCCH3NHCH(COO)2)CH2(COO)2(H2O)2}– при 594.7 а.е.м. (вычислено 595.1). Полученные данные хорошо согласуются с результатами элементного анализа.
Таблица 1.
Кристаллографические данные и параметры уточнения структуры
| Соединение | (NBu4)[2-B10H9NHC(CH3)NHCH(COOEt)2] |
|---|---|
| Эмпирическая формула | C25H61B10N3O4 |
| М | 575.9 |
| T, K | 100(2) |
| Сингония | Моноклинная |
| Пр. гр. | P21/n |
| a, Å | 14.5711(6) |
| b, Å | 13.9611(5) |
| c, Å | 18.0215(7) |
| α, град | 90 |
| β, град | 102.666(3) |
| γ, град | 90 |
| V, Å3 | 3576.87 |
| Z | 4 |
| ρx, мг/м3 | 1.069 |
| μ, мм–1 | 0.065 |
| Размер кристалла, мм | 0.200 × 0.180 × 0.060 |
| Интервал θ, град | 3.20–27.25 |
| Общее число рефлексов | 37 955 |
| независимых (N) | 8165 |
| [Rint], | [0.1225] |
| в том числе с I > 2σ(I) (N0) | 4011 |
| Tmax, Tmin | – |
| Отражения/ограничения/параметры | 8165/0/412 |
| GOOF (F2) | 1.047 |
| R1, wR2 для N0 | 0.1020, 0.1742 |
| R1, wR2 для N | 0.3088, 0.2496 |
| Δρmax/Δρmin, e/Å3 | 0.743/–0.571 |
Список литературы
Zhdanov A.P., Retivov V.M., Razgonyaeva G.A. et al. // Russ. J. Inorg. Chem. 2015. V. 60. P. 771. [Жданов А.П., Ретивов В.М., Разгоняева Г.А. и др. // Журн. неорган. химии. 2015. Т. 60. С. 852.]https://doi.org/10.1134/S0036023615070189
Shakirova O.G., Lavrenova L.G., Bogomyakov A.S. et al. // Russ. J. Inorg. Chem. 2015. V. 60. P. 786. [Шакирова О.Г., Лавренова Л.Г., Богомяков А.С. и др. // Журн. неорган. химии. 2015. Т. 60. С. 869.]https://doi.org/10.1134/S003602361507013X
Malinina E.A., Kochneva I.K., Polyakova I.N. et al. // Inorg. Chim. Acta. 2018. V. 479. P. 249. https://doi.org/10.1016/j.ica.2018.04.059
Kaszynski P., Huang J., Jenkins G.S. et al. // Mol. Cryst. Liq. Cryst. Sci. Technol., Sect. A. 1995. V. 260. P. 315. https://doi.org/10.1080/10587259508038705
Plekhanov A.I., Markov R.V., Rautian S.G. et al. // Proc. SPIE-The Int. Soc. Opt. Eng. 1998. P. 20. https://doi.org/10.1117/12.328187
Jankowiak A., Baliński A., Harvey J.E. et al. // J. Mater. Chem. 2013. V. 1. P. 1144. https://doi.org/10.1039/c2tc00547f
Bregadze V., Sivaev I. // Boron Sci., CRC Press. 2011. P. 181. https://doi.org/10.1201/b11199-14
Mrugala M.M. // Discov. Med. V. 15. P. 221. https://www.discoverymedicine.com/Maciej-M-Mrugala/2013/04/25/advances-and-challenges-in-the-treatment-of-glioblastoma-a-clinicians-perspective
Nedunchezhian K., Aswath N., Thiruppathy M. et al. // J. Clin. Diagnostic Res. 2016. V. 10. ZE01. https://doi.org/10.7860/JCDR/2016/19890.9024
Bavarnegin E., Kasesaz Y., Wagner F.M. // J. Instrum. 2017. V. 12. P05005. https://doi.org/10.1088/1748-0221/12/05/P05005
Nakamura S., Igaki H., Okamoto H. et al. // Phys. Medica. 2019. V. 58. P. 121. https://doi.org/10.1016/j.ejmp.2019.02.010
Semioshkin A., Laskova J., Wojtczak B. et al. // J. Organomet. Chem. 2009. V. 694. P. 1375. https://doi.org/10.1016/j.jorganchem.2008.12.024
Matveev E.Y., Razgonyaeva G.A., Mustyatsa V.N. et al. // Russ. Chem. Bull. 2010. V. 59. P. 556. https://doi.org/10.1007/s11172-010-0125-0
Prikaznov A.V., Shmal’ko A.V., Sivaev I.B. et al. // Polyhedron. 2011. V. 30. P. 1494. https://doi.org/10.1016/j.poly.2011.02.055
Goszczyński T.M., Kowalski K., Leśnikowski Z.J. // Biochim. Biophys. Acta. 2015. V. 1850. P. 411. https://doi.org/10.1016/j.bbagen.2014.11.015
Preetz W., Nachtigal C. // Z. Anorg. Allg. Chem. 1995. V. 621. P. 1632. https://doi.org/10.1002/zaac.19956211003
El Anwar S., Holub J., Tok O. et al. // J. Organomet. Chem. 2018. V. 865. P. 189. https://doi.org/10.1016/j.jorganchem.2018.02.050
Jasper S.A., Jones R.B., Mattern J. et al. // Inorg. Chem. 1994. V. 33. P. 5620. https://doi.org/10.1021/ic00103a005
Naoufal D., Grüner B., Bonnetot B. et al. // Polyhedron. 1999. V. 18. P. 931. https://doi.org/10.1016/S0277-5387(98)00354-4
Naoufal D., Assi Z., Abdelhai E. et al. // Inorg. Chim. Acta. 2012. V. 383. P. 33. https://doi.org/10.1016/j.ica.2011.10.033
Kaszyński P., Ringstrand B. // Angew. Chemie Int. Ed. 2015. V. 54. P. 6576. https://doi.org/10.1002/anie.201411858
Shelly K., Knobler C.B., Hawthorne M.F. // Inorg. Chem. 1992. V. 31. P. 2889. https://doi.org/10.1021/ic00039a041
Zhdanov A.P., Lisovsky M.V., Goeva L.V. et al. // Russ. Chem. Bull. 2009. V. 58. P. 1694. https://doi.org/10.1007/s11172-009-0234-9
Zhdanov A.P., Polyakova I.N., Razgonyaeva G.A. et al. // Russ. J. Inorg. Chem. 2011. V. 56. P. 847. https://doi.org/10.1134/S003602361106026X
Burianova V.K., Mikherdov A.S., Bolotin D.S. et al. // J. Organomet. Chem. 2018. P. 870. https://doi.org/10.1016/j.jorganchem.2018.06.017
Daines E.A., Bolotin D.S., Bokach N.A. et al. // Inorg. Chim. Acta. 2018. V. 471. P. 372. https://doi.org/10.1016/j.ica.2017.11.054
Mindich A.L., Bokach N.A., Kuznetsov M.L. et al. // Chempluschem. 2012. P. 77. https://doi.org/10.1002/cplu.201200257
Zhdanova K.A., Zhdanov A.P., Ezhov A.V. et al. // Russ. Chem. Bull. 2014. V. 63. P. 194. [Жданова К.А., Жданов А.П., Ежов А.В. и др. // Изв. АН. 2014. Т. 63. С. 194.]https://doi.org/10.1007/s11172-014-0413-1
Losytskyy M.Y., Kovalska V.B., Varzatskii O.A. et al. // J. Lumin. 2016. V. 169. P. 51. https://doi.org/10.1016/j.jlumin.2015.08.042
Bolotin D.S., Burianova V.K., Novikov A.S. et al. // Organometallics. 2016. P. 35. https://doi.org/10.1021/acs.organomet.6b00678
Zhdanov A.P., Klyukin I.N., Bykov A.Y. et al. // Polyhedron. 2017. V. 123. P. 176. https://doi.org/10.1016/j.poly.2016.11.035
Klyukin I.N., Zhdanov A.P., Bykov A.Y. et al. // Russ. J. Inorg. Chem. 2017. V. 62. P. 1479. [Клюкин И.Н., Жданов А.П., Быков А.Ю. и др. // Журн. неорган. химии. 2017. V. 62. P. 1486.]https://doi.org/10.1134/S0036023617110109
Butterworth K.T., McMahon S.J., Currell F.J. et al. // Nanoscale. 2012. V. 4. P. 4830. https://doi.org/10.1039/c2nr31227a
Butterworth K.T., McMahon S.J., Taggart L.E. // Centre, Transl. Cancer Res. 2013. V. 2. P. 269. https://doi.org/10.3978/j.issn.2218-676X.2013.08.03
Haume K., Rosa S., Grellet S. et al. // Cancer Nanotechnol. 2016. V. 7. P. 8. https://doi.org/10.1186/s12645-016-0021-x
Lux F., Sancey L., Bianchi A. et al. // Nanomedicine. 2015. V. 10. P. 1801. https://doi.org/10.2217/nnm.15.30
Hainfeld J.F., Slatkin D.N., Smilowitz H.M. // Phys. Med. Biol. 2004. V. 49. P. N309. https://doi.org/10.1088/0031-9155/49/18/N03
Hainfeld J.F., Smilowitz H.M., O’Connor M.J. et al. // Nanomedicine. 2013. V. 8. P. 1601. https://doi.org/10.2217/nnm.12.165
Al Zaki A., Joh D., Cheng Z. et al. // ACS Nano 2014. V. 8. P. 104. https://doi.org/10.1021/nn405701q
McQuade C., Al Zaki A., Desai Y. et al. // Small. 2015. V. 11. P. 834. https://doi.org/10.1002/smll.201401927
Choi G.-H., Seo S.-J., Kim K.-H. et al. // Radiat. Oncol. 2012. V. 7. P. 184. https://doi.org/10.1186/1748-717X-7-184
Maggiorella L., Barouch G., Devaux C. et al. // Futur. Oncol. 2012. V. 8. P. 1167. https://doi.org/10.2217/fon.12.96
SAINT. Version 7.23A. Bruker AXS Inc. Madison, Wisconsin, USA. 2003.
SADABS-2004/1. Bruker AXS Inc. Madison, Wisconsin, USA. 2004.
Sheldrick G.M. // Acta Crystallogr. 2008. V. 64. P. 112. https://doi.org/10.1107/S0108767307043930
Sheldrick G.M. // Acta Crystallogr., Sect. A: Found. Crystallogr. 2015. V. 71. P. 3. https://doi.org/10.1107/S2053273314026370
Sivaev I.B., Votinova N.A., Bragin V.I. et al. // J. Organomet. Chem. 2002. V. 657. P. 163. https://doi.org/10.1016/S0022-328X(02)01419-5
Dolomanov O.V., Bourhis L.J., Gildea R.J. et al. // J. Appl. Crystallogr. 2009. V. 42. P. 339. https://doi.org/10.1107/S0021889808042726
Allen F.H., Kennard O., Watson D.G. et al. // J. Chem. Soc., Perkin Trans. 2. 1987. S1. https://doi.org/10.1039/p298700000s1
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии