

Журнал неорганической химии, 2020, T. 65, № 4, стр. 435-442
Формирование одномерных иерархических наноструктур MoO3 в гидротермальных условиях
Т. Л. Симоненко a, *, В. А. Бочарова a, b, Н. П. Симоненко a, Ф. Ю. Горобцов a, Е. П. Симоненко a, А. Г. Мурадова b, В. Г. Севастьянов a, Н. Т. Кузнецов a
a Институт общей и неорганической химии им. Н.С. Курнакова РАН
119991 Москва, Ленинский пр-т, 31, Россия
b Российский химико-технологический университет им. Д.И. Менделеева
125047 Москва, Миусская пл., 9, Россия
* E-mail: egorova.offver@gmail.com
Поступила в редакцию 20.11.2019
После доработки 29.11.2019
Принята к публикации 02.12.2019
Аннотация
С помощью гидротермального метода получены одномерные структуры h-MoO3. Показано влияние условий гидротермальной обработки на микроструктуру и дисперсность формирующегося оксидного порошка. С помощью рентгенофазового анализа подтверждено получение гексагональной структуры и отсутствие кристаллических примесей. Методами сканирующей емкостной и Кельвин-зондовой силовой микроскопии оценены электрофизические характеристики поверхности полученного порошка. Исследована кинетика фотокаталитического разложения Родамина Б под воздействием видимого излучения в присутствии h-MoO3.
ВВЕДЕНИЕ
Оксид молибдена(VI) представляет собой полупроводник n-типа с шириной запрещенной зоны 2.8–3.6 эВ и благодаря своей окислительно-восстановительной активности находит широкое применение в газовой сенсорике [1–3], альтернативной энергетике (в частности, при разработке литий-ионных батарей [4, 5] и суперконденсаторов [6, 7]), катализе [8–10], оптике [11], устройствах хранения информации [12]. Как известно, MoO3 может существовать в нескольких полиморфных модификациях: термодинамически устойчивой форме α-MoO3 (Pnma) и метастабильных β-MoO3 (Р21/с), ε-MoO3 (Р21/m), а также h-MoO3 (Р63/m) [9, 13, 14]. Несмотря на то, что кристаллическая решетка всех перечисленных разновидностей оксида молибдена(VI) сформирована из октаэдров [MoO6], их расположение друг относительно друга различно, что во многом определяет особые свойства каждой из модификаций. Так, для наиболее изученной из них α-MoO3 характерно формирование слоистых анизотропных структур, вытянутых вдоль направления [010], что обусловлено расположением кислород-молибденовых октаэдров в виде слоев, образующих орторомбическую структуру кристаллов. Тогда как гексагональная структура оксида молибдена(VI) образована зигзагообразными цепочками октаэдров [MoO6], связанных друг с другом в цис-позициях, формирующих обширные одномерные каналы вдоль направления [001]. Среди перечисленных кристаллических структур устойчивая модификация α-MoO3 достаточно хорошо изучена, а метастабильная гексагональная модификация h-MoO3 исследована в меньшей степени. При этом в литературе отмечается, что особый интерес к изучению метастабильных фаз обусловлен тем, что по сравнению с устойчивыми полиморфными модификациями рассматриваемого вещества они могут обладать уникальными свойствами [15–18].
Известно большое число работ по получению орторомбической структуры оксида молибдена(VI) с различной морфологией (наностержни [19], наноленты [20, 21], нанотрубки [22], нанолисты [23, 24], нановолокна [25], полые сферы [26] и т.п.) с использованием целого ряда синтетических методов и подходов, в частности, с помощью спрей-пиролиза [27, 28], физического осаждения из газовой фазы [29], магнетронного распыления [30], гидротермального метода [21]. Получение одномерных структур гексагонального h-MoO3 требует более тщательного подбора метода и условий синтеза в силу меньшей термодинамической устойчивости, а в литературе имеется небольшое количество публикаций, сообщающих об успешном получении данного материала [31–34]. При этом авторам зачастую не удавалась достичь необходимой однородности и иерархической организации получаемых структур, что в значительной степени влияло на функциональные свойства продукта [7, 16, 35]. Наиболее перспективными в данном контексте методами синтеза будут совместное осаждение гидроксидов металлов и гидротермальный метод, позволяющие в широких пределах варьировать условия синтеза и тем самым тонко контролировать дисперсность, фазовый состав и микроструктуру получаемых наноматериалов [36–40].
Таким образом, целью данной работы является изучение процесса синтеза одномерных структур h-MoO3 с помощью гидротермального метода, а также исследование микроструктуры, электрофизических и фотокаталитических свойств получаемого порошка.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Одномерные структуры гексагонального оксида молибдена(VI) получали с использованием в качестве исходного реагента парамолибдата аммония (NH4)6Mo7O24 ⋅ 4H2O марки “х. ч.”, водный раствор которого доводили до целевого значения рН 1.5 путем добавления водных растворов триэтаноламина (φ = 10%) и азотной кислоты (φ = 30%). Далее полученную реакционную систему переносили в тефлоновый автоклав и подвергали гидротермальной обработке при температуре 160°С в течение 2 ч. Образовавшуюся в ходе синтеза дисперсную фазу отделяли и промывали дистиллированной водой путем циклического центрифугирования, после чего проводили ее сушку при 100°С в течение 3 ч.
Синхронный (ТГА/ДСК) термический анализ порошков, полученных в результате гидротермального синтеза и последующей сушки при 100°С, был выполнен с использованием термоанализатора SDT Q-600 (контролируемый нагрев проводили в Al2O3-микротиглях в интервале температур 20–820°С со скоростью 10 град/мин в токе воздуха, скорость потока газа составляла 250 мл/мин, масса навески – 8.57 мг).
Рентгенофазовый анализ (РФА) порошков выполняли на рентгеновском дифрактометре Bruker D8 Advance (CuKα-излучение, 2θ = 5°–80°, разрешение 0.02°, время накопления сигнала в точке 0.3 с).
Для записи ИК-спектров пропускания порошков были приготовлены их суспензии в вазелиновом масле, которые в виде пленки помещали между стеклами KBr. Спектральный анализ проводили в диапазоне волновых чисел 350–4000 см–1 (время накопления сигнала составляло 15 с, разрешение – 1 см–1) с использованием ИК-Фурье-спектрометра ИнфраЛЮМ ФТ-08.
Микроструктуру полученных порошков изучали с помощью растровой электронной микроскопии (РЭМ) на трехлучевой рабочей станции NVision 40 (Carl Zeiss), оснащенной энергодисперсионной приставкой-микроанализатором INCA X-MAX (Oxford Instruments).
Исследование рельефа поверхности полученных порошков с помощью атомно-силовой микроскопии (АСМ), а также изучение их электрофизических свойств методами сканирующей емкостной микроскопии (СЕМ) и Кельвин-зондовой силовой микроскопии (КЗСМ) проводили на приборе NT-MDT Solver-Pro с использованием зондов с алмазными иглами DCP-20. Сканирование проводили в полуконтактном режиме.
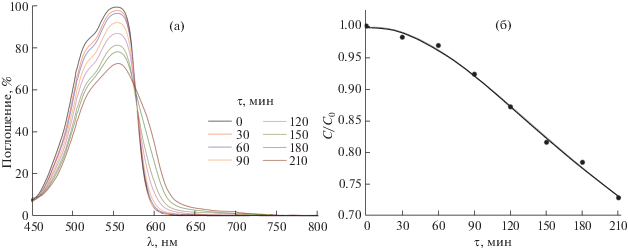
Фотокаталитическую активность полученных одномерных структур h-MoO3 исследовали по кинетике реакции фоторазложения органического водорастворимого красителя (Родамина Б) под действием видимого излучения. Раствор красителя (10 мг/л), содержащий 0.01 г порошка оксида молибдена(VI), подвергали облучению от источника мощностью 100 Вт при перемешивании в течение 210 мин. Отбор пробы исследуемого раствора проводили каждые 30 мин и исследовали процесс фотодеградации по изменению величины поглощения с помощью UV-Vis спектрофотометра СФ-56. Изменение концентрации Родамина Б оценивали по величине интенсивности его полосы поглощения с максимумом при 554 нм.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Как видно из ИК-спектра пропускания порошка оксида молибдена(VI), полученного после проведения гидротермального синтеза и сушки (рис. 1), полосы с максимумами при 3493 см–1 (валентные колебания OH-групп) и 1610 см–1 (деформационные колебания НОН) свидетельствуют о наличии абсорбированной воды в структуре порошка. Полосы с максимумами при 3192 и 1667 см–1 соответствуют валентным и деформационным колебаниям связей N–H, указывающим на остаточное содержание ионов ${\text{NH}}_{4}^{ + },$ которые могут выступать в роли стабилизатора гексагональной структуры MoO3 [17, 41]. Кроме того, в представленном спектре присутствует пять характеристических полос, присущих h-MoO3, с максимумами при 978 и 910 см–1 (валентные колебания двойных связей Mo=O), 721 и 522 см–1 (асимметричные и симметричные валентные колебания функциональных групп Mo–O–Mo соответственно), а также полоса с максимумом при 592 см–1 (деформационные колебания Mo–O) [8, 42]. Необходимо также отметить, что отсутствие в спектре полос поглощения с максимумами при 1000, 867 и 553 см–1, характерных для α-модификации оксида молибдена(VI), дополнительно подтверждает формирование целевого оксида h-MoO3.
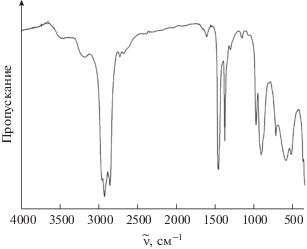
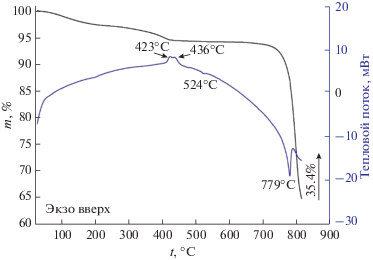
На следующем этапе с помощью синхронного (ТГА/ДСК) термического анализа в токе воздуха (250 мл/мин) в интервале температур 20–820°С (скорость нагрева составляла 10 град/мин) было изучено термическое поведение полученного порошка h-MoO3. Как видно из термограммы (рис. 2), при нагревании исследуемого соединения наблюдаются четыре ступени потери массы в интервалах температур 20–230, 230–440, 440–680 и 680–820°С. Первая (∼2.8%) из них сопровождается незначительным пологим эндотермическим эффектом с максимумом при 210°С, обусловленным удалением остаточного растворителя; вторая ступень (∼2.8%), которой соответствует экзотермический эффект с максимумом при 420°С, вероятно, связана с окислением органических продуктов разложения триэтаноламина. Экзотермический эффект с максимумом при 434°С без потери массы может быть отнесен к фазовому переходу метастабильной полиморфной модификации h-MoO3 в устойчивую структуру α-MoO3 [17]. При температуре ∼524°С имеет место малоинтенсивный экзоэффект, который сопровождается незначительной потерей массы (∼0.4%) и, согласно [43], может быть обусловлен удалением из структуры соединения интеркалированных ионов ${\text{NH}}_{4}^{ + },$ а также структурно связанных молекул воды. Четвертая ступень потери массы (29.4%) в интервале 680–800°С, которая сопровождается интенсивным экзотермическим эффектом с максимумом при 780°С, связана с протеканием процесса плавления исследуемого соединения.
Рис. 2.
Кривые ТГА (черная) и ДСК (синяя) в потоке воздуха для порошка h-MoO3, полученного в результате гидротермального синтеза и последующей сушки.
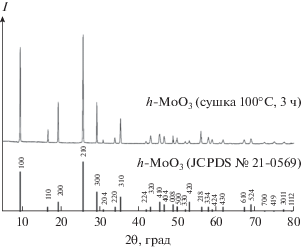
Кристаллическая структура полученного вещества была исследована с помощью РФА, результаты которого хорошо согласуются с данными ИК-спектроскопии и подтверждают формирование целевой гексагональной структуры оксида молибдена(VI) с пр. гр. Р63/m (рис. 3). Как видно из рентгенограммы, полученный порошок также не содержит посторонних фаз либо кристаллических примесей. Согласно литературным источникам [7, 41], формированию гексагональной модификации MoO3 в гидротермальных условиях в основном способствуют такие факторы, как низкая концентрация кислоты, наличие водной среды, относительно низкая температура и небольшая длительность термообработки. Кристаллическая структура h-MoO3 образуется за счет самоорганизации октаэдров [MoO6] при участии катионов ${\text{NH}}_{4}^{ + }$ и молекул воды, выполняющих роль структурообразователей, способствуя росту кристаллитов преимущественно вдоль направления с, что приводит к формированию стержневидных структур, которые также удалось зафиксировать с помощью растровой электронной микроскопии (рис. 4). Как видно из микрофотографии, порошок h-MoO3 состоит из вытянутых стержней с четкими правильными гранями, средний диаметр которых составляет ∼8 мкм, а отношение длины к диаметру ∼3.3. Поверхность стержней является достаточно однородной и состоит из наноразмерных частиц со средним размером ∼30 нм. Изучение микроструктуры порошка в режиме контраста по среднему атомному номеру (детектор отраженных электронов) и результаты энергодисперсионного элементного микроанализа свидетельствуют о формировании однородного продукта целевого состава.

Микроструктура поверхности полученных стержней оксида молибдена также исследована с помощью атомно-силовой микроскопии. Как видно на рис. 5а, грань исследуемого стержня достаточно гладкая – перепад высот составляет около 70 нм. Частицы, составляющие стержень, достаточно плотно прилегают друг к другу, каких-либо пор на поверхности не обнаружено. В целом результаты АСМ хорошо согласуются с микрофотографиями РЭМ.
Рис. 5.
Рельеф поверхности стержня h-MoO3 по данным АСМ (а), результаты СЕМ (б) и распределение поверхностного потенциала на участке стержня h-MoO3 за вычетом базовой кривой (в). Зависимость ∂C/∂Z от расстояния Z между зондом и поверхностью стержня h-MoO3 (г).
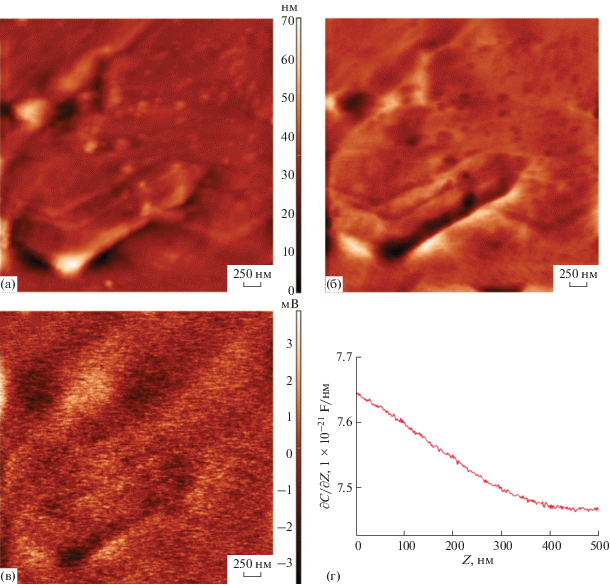
С использованием сканирующей емкостной и Кельвин-зондовой микроскопии исследованы электрофизические свойства поверхности стержней h-MoO3. Как видно из результатов, приведенных на рис. 5б и 5в, поверхность стержня достаточно однородна по своим свойствам: она имеет практически постоянный потенциал (рис. 5в), при этом емкостной контраст также невелик, в основном проявляются эффекты, связанные с рельефом поверхности.
На основании данных Кельвин-зондовой силовой микроскопии была рассчитана работа выхода электрона с поверхности стержня оксида молибдена по формуле:
С помощью атомно-силовой микроскопии была также определена локальная электрическая емкость на поверхности стержня оксида молибдена. В ходе измерений в режиме cканирующей емкостной микроскопии запись сигнала проводилась на частоте второй гармоники 2ω, при которой сила взаимодействия кончика кантилевера и образца равняется:
(1)
${{F}_{{2\omega }}} = \,\,~\frac{1}{4}U_{1}^{2}\frac{{\partial C}}{{\partial Z}}{\text{cos}}\left( {2\omega t} \right),$Следовательно, из уравнения (2), зная величину амплитуды, можно вычислить ∂C/∂Z. Для определения емкости требуется взять интеграл, что легко сделать графически: фиксируется изменение амплитуды колебаний в зависимости от высоты зонда над точкой поверхности, амплитуда переводится в градиент ∂C/∂Z и далее рассчитывается площадь под полученным спектром. Типичный результат такой спектроскопии на второй гармонической частоте приведен на рис. 5г. По результатам данной спектроскопии, среднее значение емкости для поверхности стержня оксида молибдена равняется 3.8 × 10–18 Ф, что в пересчете на единицу площади соответствует удельному значению 1.01 пФ/м2.
Фотокаталитические свойства полученного порошка оценивались по методу фотодеградации красителя. В нашем случае в качестве модельного красящего вещества был выбран Родамин Б, поскольку он входит в состав выбросов текстильной и пищевой промышленности, являясь достаточно токсичным канцерогеном, поэтому исследование процесса его разложения в водных средах представляет также и практический интерес.
Как видно из рис. 6а, при увеличении времени облучения наблюдается батохромный сдвиг спектра поглощения, что свидетельствует о протекании реакций деэтилирования Родамина Б и формирования промежуточных продуктов в ходе его разложения. Уменьшение интенсивности полосы поглощения с максимумом при 554 нм во времени связано с процессом обесцвечивания данного красителя вследствие разрушения его хромофорных ароматических колец [45]. Таким образом, согласно полученным экспериментальным данным (рис. 6б), в ходе процесса облучения раствора красителя, содержащего в качестве катализатора полученный порошок h-MoO3, удалось достичь снижения концентрации Родамина Б на 30%, что представляет собой достаточно существенный результат с учетом небольшой мощности источника видимого излучения.
ЗАКЛЮЧЕНИЕ
В ходе данной работы изучен процесс гидротермального синтеза одномерных иерархических структур h-MoO3. Установлено, что используемый метод и условия синтеза позволяют получать однофазный целевой продукт, характеризующийся гексагональной кристаллической структурой Р63/m. Изучение микроструктуры поверхности полученного порошка в режиме контраста по среднему атомному номеру также подтвердило однородность материала и отсутствие примесей другого химического состава.
Исследованы электрофизические свойства поверхности полученного материала, в частности, рассчитана работа выхода электрона с поверхности (4.91 эВ), а также величина удельной емкости (1.01 пФ/м2).
По результатам оценки фотокаталитической активности стержней h-MoO3 установлено, что полученный порошок демонстрирует значительную каталитическую активность при разложении Родамина Б под воздействием излучения в видимой области спектра.
Таким образом, полученные иерархические структуры гексагонального оксида молибдена(VI) по своим физико-химическим свойствам представляют значительный практический интерес и могут быть использованы в качестве материала электродов суперконденсаторов, а также фотокатализаторов, в частности для очистки воды.
Список литературы
Mo Y., Tan Z., Sun L. et al. // J. Alloys Compd. 2020. V. 812. P. 152166. https://doi.org/10.1016/j.jallcom.2019.152166
Sui L.-I., Xu Y.-M., Zhang X.-F. et al. // Sens Actuators B. 2015. V. 208. P. 406. https://doi.org/10.1016/j.snb.2014.10.138
Bai S., Chen S., Chen L. et al. // Sens Actuators B. 2012. V. 174. P. 51. https://doi.org/10.1016/j.snb.2012.08.015
Wang Z., Madhavi S., Lou X.W. // J. Phys. Chem. C. 2012. V. 116. P. 12508. https://doi.org/10.1021/jp304216z
Hu X., Zhang W., Liu X. et al. // Chem. Soc. Rev. 2015. V. 44. P. 2376. https://doi.org/10.1039/c4cs00350k
Sangeetha D.N., Krishna Bhat D., Selvakumar M. // Ionics. 2019. V. 25. P. 607. https://doi.org/10.1007/s11581-018-2684-2
Sangeetha D.N., Sowmya Holla R., Ramachandra Bhat B. et al. // Int. J. Hydrogen Energ. 2019. https://doi.org/10.1016/j.ijhydene.2019.10.029
Chithambararaj A., Sanjini N.S., Chandra Bose A., Velmathi S. // Catal. Sci. Technol. 2013. V. 3. P. 1405. https://doi.org/10.1039/C3CY20764A
Pan W., Tian R., Jin H. et al. // Chem. Mater. 2010. V. 22. P. 6202. https://doi.org/10.1021/cm102703s
Chithambararaj A., Sanjini N.S., Velmathi S., Chandra Bose A. // Phys Chem. Chem. Phys. 2013. V. 15. P. 14 761. https://doi.org/10.1039/c3cp51796a
Zheng L., Xu Y., Jin d., Xie Y. // Chem. Mater. 2009. V. 21. P. 5681. https://doi.org/10.1021/cm9023887
Wei G., Qin W., Zhang D. // J. Alloys Compd. 2009. V. 481. P. 417. https://doi.org/10.1016/j.jallcom.2009.03.007
Atuchin V.V., Gavrilova T.A., Kostrovsky V.G. et al. // Inorg. Mater. 2008. V. 44. P. 622. https://doi.org/10.1134/S0020168508060149
Song Y., Zhao Y., Huang Z., Zhao J. // J. Alloys Compd. 2017. V. 693. P. 1290. https://doi.org/10.1016/j.jallcom.2016.10.092
Lu X., Wang R., Yang F. et al. // J. Mater. Chem. C. 2016. V. 4. P. 6720. https://doi.org/10.1039/C6TC01656A
Paraguay-Delgado F., Mendoza Duarte M.E., Kalu O. et al. // J. Therm. Anal. Calorim. 2019. https://doi.org/10.1007/s10973-019-08842-0
Liu Y., Yang S., Lu Y. et al. // Appl. Surf. Sci. 2015. V. 359. P. 114. https://doi.org/10.1016/j.apsusc.2015.10.071
Hu H., Deng C., Xu J. et al. // J. Exp. Nanosci. 2015. V. 10. P. 1336. https://doi.org/10.1080/17458080.2015.1012654
Chen J.-S., Cheah Y.-L., Madhavi S., Lou X.W. // J. Phys. Chem. C. 2010. V. 114. P. 8675. https://doi.org/10.1021/jp1017482
Chu W.G., Zhang L.N., Wang H.F. et al. // J. Mater. Res. 2007. V. 22. P. 1609. https://doi.org/10.1557/JMR.2007.0217
Gao B., Fan H., Zhang X. // J. Phys. Chem. Solids. 2012. V. 73. P. 423. https://doi.org/10.1016/j.jpcs.2011.11.019
Hu S., Wang X. // J. Am. Chem. Soc. 2008. V. 130. P. 8126. https://doi.org/10.1021/ja801448c
Bai H.X., Liu X.H., Zhang Y.C. // Mater. Lett. 2009. V. 63. P. 100. https://doi.org/10.1016/j.matlet.2008.09.016
Kalantar-Zadeh K., Tang J., Wang M. et al. // Nanoscale. 2010. V. 2. P. 429. https://doi.org/10.1039/b9nr00320g
Dewagan K., Sinha N.N., Sharma P.K. et al. // Cryst-EngComm. 2011. V. 13. P. 927. https://doi.org/10.1039/c0ce00271b
Wang Z., Wang H., Yang C., Wu J. // Mater. Lett. 2010. V. 64. P. 2170. https://doi.org/10.1016/j.matlet.2010.07.035
Pandeeswari R., Jeyaprakash B.G. // Biosens. Bioelectron. 2014. V. 53. P. 182. https://doi.org/10.1016/j.bios.2013.09.057
Díaz C., Lavayen V., O’Dwyer C. // J. Solid State Chem. 2010. V. 183. P. 1595. https://doi.org/10.1016/j.jssc.2010.05.006
Wang K.-K., Wang F.-X., Liu Y.-D., Pan G.-B. // Mater. Lett. 2013. V. 102–103. P. 8. https://doi.org/10.1016/j.matlet.2013.03.092
Navas I., Vinodkumar R., Lethy K.J. et al. // J. Phys. D. Appl. Phys. 2009. V. 42. P. 175305. https://doi.org/10.1088/0022-3727/42/17/175305
Sun T., Xu L., Wei S. et al. // Int. J. Refract. Met. H. 2020. V. 86. P. 105085. https://doi.org/10.1016/j.ijrmhm.2019.105085
Xu Y., Zhang X., Li Y. et al. // Mater. Lett. 2018. V. 210. P. 314. https://doi.org/10.1016/j.matlet.2017.09.010
Ama O.M., Kumar N., Adams F.V., Ray S.S. // Electrocatalysis. 2018. V. 9. P. 623. https://doi.org/10.1007/s12678-018-0471-5
Almodóvar P., Díaz-Guerra C., Ramírez-Castellanos J., González-Calbet J.M. // CrystEngComm. 2018. V. 20. P. 4954. https://doi.org/10.1039/C8CE00747K
Yogananda H.S., Nagabhushana H., Darshan G.P. et al. // J. Alloys Compd. 2018. V. 745. P. 874. https://doi.org/10.1016/j.jallcom.2017.11.278
Simonenko T.L., Ivanova V.M., Simonenko N.P. et al. // Russ. J. Inorg. Chem. 2019. V. 64. https://doi.org/10.1134/S0036023619140080
Simonenko T.L., Simonenko N.P., Simonenko E.P. et al. // Russ. J. Inorg. Chem. 2019. V. 64. P. 1475. https://doi.org/10.1134/S0036023619120167
Egorova T.L., Kalinina M.V., Simonenko E.P. et al. // Russ. J. Inorg. Chem. 2017. V. 62. P. 1275. https://doi.org/10.1134/S0036023617100072
Simonenko T.L., Kalinina M.V., Simonenko N.P. et al. // Glass. Phys. Chem. 2018. V. 44. P. 314. https://doi.org/10.1134/S1087659618040144
Simonenko T.L., Kalinina M.V., Simonenko N.P. et al. // Ceram. Int. 2018. V. 44. P. 19879. https://doi.org/10.1016/j.ceramint.2018.07.249
Zakharova G.S., Schmidt C., Ottman A. et al. // J. Solid State Electrochem. 2018. V. 22. P. 3651. https://doi.org/10.1007/s10008-018-4073-1
Wongkrua P., Thongtem T., Thongtem S. // J. Nanomater. 2013. P. 702679. https://doi.org/10.1155/2013/702679
Chithambararaj A., Bhagya Mathi D., Rajeswari Yogamalar N., Chandra Bose A. // Mater. Res. Express. 2015. V. 2. P. 055004. https://doi.org/10.1088/2053-1591/2/5/055004
Vasilopoulou M., Douvas A.M., Georgiadou D.G. et al. // J. Am. Chem. Soc. 2012. V. 134. P. 16178. https://doi.org/10.1021/ja3026906
Alex K.V., Prabhakaran A., Jayakrishnan A.R. et al. // ACS Appl. Mater. Interfaces. 2019. V. 11. P. 40114. https://doi.org/10.1021/acsami.9b14919
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии