

Журнал неорганической химии, 2020, T. 65, № 4, стр. 467-474
Селективный гидротермальный синтез [(CH3)2NH2]V3O7, VO2(D) и V2O3 в присутствии N,N-диметилформамида
Л. А. Носикова a, А. Е. Баранчиков b, c, *, А. Д. Япрынцев c, О. С. Иванова c, М. А. Теплоногова b, c, В. К. Иванов b, c
a МИРЭА – Российский технологический университет (Институт тонких химических технологий
им. М.В. Ломоносова)
119571 Москва, пр-т Вернадского, 86, Россия
b Московский государственный университет им. М.В. Ломоносова
119991 Москва, ул. Ленинские горы, 1, Россия
c Институт общей и неорганической химии им. Н.С. Курнакова РАН
119991 Москва, Ленинский пр-т, 31, Россия
* E-mail: a.baranchikov@yandex.ru
Поступила в редакцию 28.10.2019
После доработки 05.11.2019
Принята к публикации 08.11.2019
Аннотация
Исследовано взаимодействие пентаоксида диванадия и диметилформамида в гидротермальных условиях при температурах 180–250°С. Показано, что в зависимости от температуры и продолжительности (1–7 сут) гидротермального синтеза взаимодействие V2O5 и (CH3)2N–CHO приводит к получению однофазных порошков [(CH3)2NH2]V3O7, VO2(D) и V2O3 с выходом до 50–85%. Варьирование стартового значения рН реакционных смесей в диапазоне 0.5–3.7 и мольного соотношения V2O5 : (CH3)2N–CHO в диапазоне 1 : 5–1 : 50 не оказывает существенного влияния на фазовый состав продуктов синтеза.
ВВЕДЕНИЕ
Оксидные соединения ванадия могут служить основой для получения неорганических материалов, обладающих интересными оптическими и электрофизическими свойствами, что позволяет использовать их для создания современных источников постоянного тока, электрохромных и термохромных покрытий, газовых сенсоров, фотоэлектрохимических преобразователей и др. [1–10]. Например, для диоксида (VO2) и сесквиоксида (V2O3) ванадия характерен фазовый переход металл–полупроводник, который сопровождается значительным изменением оптических и электрофизических характеристик [11–14].
Существенное влияние на физические характеристики материалов на основе оксидов ванадия, включая величину электропроводности, показатель преломления и ширину фотонной запрещенной зоны, оказывает их химический и фазовый состав [15]. Система V–O является достаточно сложной, и в ней возможно существование оксидов различного химического состава, в которых ванадий проявляет как целочисленные, так и переменные степени окисления, например, в так называемых фазах Уодсли и фазах Магнели [1, 2, 16]. Кроме того, для ряда оксидов ванадия характерно наличие нескольких полиморфных модификаций, которые могут существовать при обычных условиях в стабильном или метастабильном состоянии. Так, диоксид ванадия имеет минимум пять полиморфных модификаций: VO2(A) [17], VO2(B) [18], VO2(C) [19], VO2(D) [20], VO2(P) [21]. Для сесквиоксида ванадия к настоящему времени получены три модификации. Стабильной при обычных условиях является V2O3 с ромбической структурой (структура корунда), при пониженных температурах (<170 K) устойчива моноклинная модификация V2O3 [22–24]. Ромбический V2O3 является парамагнетиком и характеризуется металлическим типом проводимости, моноклинный V2O3 – антиферромагнетиком и диэлектриком [24]. Одновременное изменение электропроводности и магнитных характеристик данного соединения в ходе фазового перехода может быть использовано для создания транзисторов, переключателей и запоминающих устройств. В 2011 г. была описана новая метастабильная кубическая модификация V2O3 со структурой биксбиита, проявляющая антиферромагнитные и диэлектрические свойства [25–27].
Создание материалов на основе оксидов ванадия с воспроизводимыми физическими характеристиками требует использования синтетических подходов, обеспечивающих получение продуктов с заданным фазовым составом и микроструктурой. Наиболее известным подходом является гидротермальный синтез, который обеспечивает селективное получение оксидов ванадия с высоким выходом [28]. Использование в ходе гидротермального синтеза оксидов ванадия медленно гидролизующихся соединений (метод гомогенного осаждения или метод возникающих реагентов) позволяет за счет относительно низких степеней пересыщения в реакционной системе контролировать микроструктуру продуктов синтеза и получать материалы с высокими функциональными характеристиками [29, 30]. Однако в настоящее время известно крайне мало работ, в которых описан гидротермальный синтез оксидных соединений ванадия в присутствии медленно гидролизующихся реагентов [29–31].
В качестве медленно гидролизующегося реагента, обеспечивающего селективное восстановление пентаоксида диванадия в гидротермальных условиях до различных ванадатов аммония (в частности, (NH4)2V6O16, (NH4)2V3O8, NH4V3O7), нами был предложен формамид [31]. Взаимодействие формамида и V2O5 в гидротермальных условиях при температурах до 210°С не приводит к формированию оксидов ванадия, что, по-видимому, связано с высокой устойчивостью аммонийных солей ванадиевых кислот. Аналогично формамиду N,N-диметилформамид (ДМФА) медленно гидролизуется водой [32] с образованием диметиламина и метановой кислоты, при этом скорость его гидролиза возрастает с увеличением температуры. В то же время сведения об использовании N,N-диметилформамида для синтеза оксидных соединений переходных металлов методом возникающих реагентов в гидротермальных условиях к настоящему времени отсутствуют.
Цель настоящей работы – разработка селективного метода синтеза оксидных соединений ванадия, основанного на восстановлении пентаоксида диванадия N,N-диметилформамидом в гидротермальных условиях.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В качестве исходных веществ использовали V2O5 (х. ч., ЗАО “Вектон”, Россия), N,N-диметилформамид (х. ч., ООО ТД “Химмед”), азотную кислоту (ос. ч., ООО ТД “Химмед”, Россия), дистиллированную воду. Все коммерческие реактивы применяли без дополнительной очистки.
Эксперимент проводили следующим образом. Навеску пентаоксида диванадия (91 мг) суспендировали в дистиллированной воде объемом от 5 до 30 мл, к суспензии добавляли ДМФА в количестве, соответствующем мольному соотношению V2O5 : ДМФА от 1 : 1 до 1 : 50. Кислотность среды контролировали с помощью рН-метра Crison GLP22, оснащенного универсальным электродом и термокомпенсатором. Величина pH полученных суспензий составляла от 3.6 до 3.8. В некоторых случаях для изменения pH реакционной смеси (до 0.5 и 2.0) к ней добавляли 5 М раствор азотной кислоты. Приготовленные суспензии интенсивно перемешивали на магнитной мешалке при комнатной температуре в течение 10–15 мин, затем помещали в тефлоновые автоклавы емкостью 100 мл (для проведения синтеза при температуре 180°С) или стальные автоклавы емкостью 20 мл (для проведения синтеза при температурах 210 и 250°С). Степень заполнения автоклавов составляла 35–40%. Автоклавы нагревали до 180, 210 или 250°С и выдерживали в течение 1 или 7 сут. Условия синтеза образцов приведены в табл. 1.
Таблица 1.
Условия гидротермальной обработки реакционных смесей, содержащих V2O5 и ДМФА (стартовое значение pH, температура и продолжительность); фазовый состав и выход продукта синтеза
| V2O5 : ДМФА (мольн.) | pH | t, °C | τ, сут | Продукт синтеза* | Выход**, % |
|---|---|---|---|---|---|
| 1 : 1 | ~3.7 | 180 | 1 | V2O5 · 0.5Н2О | *** |
| 1 : 10 | ~3.7 | V2O5 · 0.5Н2О | – | ||
| 1 : 50 | 0.5 | [(CH3)2NH2]V3O7 | 12 | ||
| 2.0 | [(CH3)2NH2]V3O7 | 16 | |||
| ~3.7 | [(CH3)2NH2]V3O7 | 4 | |||
| 1 : 5 | ~3.7 | 180 | 7 | [(CH3)2NH2]V3O7 | 71 |
| 1 : 10 | [(CH3)2NH2]V3O7 | 51 | |||
| 1 : 20 | [(CH3)2NH2]V3O7 | 47 | |||
| 1 : 50 | [(CH3)2NH2]V3O7 | 50 | |||
| 1 : 5 | ~3.7 | 210 | 1 | [(CH3)2NH2]V3O7 | 2 |
| 1 : 5 | ~3.7 | 210 | 7 | VO2(D) | 85 |
| 1 : 5 | ~3.7 | 250 | 1 | VO2(D) | 50 |
| 1 : 5 | ~3.7 | 250 | 7 | V2O3 | 28 |
| 1 : 10 | V2O3 | 78 |
После окончания гидротермального синтеза автоклавы охлаждали до комнатной температуры. Полученные осадки отделяли от маточных растворов декантацией (pH маточных растворов ~8) и многократно промывали дистиллированной водой с промежуточным центрифугированием (18 000 об/мин, что соответствовало ~20 000 g) в течение 10 мин. Осадки промывали до достижения электропроводности маточного раствора <0.01 мСм. Очищенные осадки сушили на воздухе при температуре 60°С в течение суток.
Термогравиметрический и дифференциальный термический анализ порошков проводили с использованием термоанализатора ТА Instruments SDT Q600 в корундовых тиглях. Масса навесок составляла 3–12 мг, нагрев до 750°С осуществляли со скоростью 10 град/мин в токе синтетического воздуха (250 см3/мин).
Рентгенофазовый анализ (РФА) проводили с использованием порошкового рентгеновского дифрактометра Bruker D8 Advance (СuKα-излучение) в диапазоне углов 2θ 5°–70° с шагом 0.02° и выдержкой не менее 0.5 с/шаг. Индицирование дифрактограмм осуществляли с помощью базы данных PDF2 (2012). Полнопрофильную обработку дифрактограмм выполняли с использованием программного обеспечения JANA2006 [33].
Микроструктуру образцов анализировали методом растровой электронной микроскопии с использованием микроскопа Саrl Zeiss NVision 40 при ускоряющем напряжении 5 кВ. Напыление электропроводящего слоя на поверхность образцов не проводили.
Анализ порошков методом ИК-спектроскопии осуществляли с помощью ИК-Фурье-спектрометра Bruker ALPHA в диапазоне частот 3000–400 см–1 в режиме нарушенного полного внутреннего отражения.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
По данным РФА, гидротермальная обработка суспензий, содержащих пентаоксид диванадия и N,N-диметилформамид, при 180°С в течение 1 сут при небольшом мольном избытке ДМФА (V2O5 : ДМФА = 1 : 1 и 1 : 10) приводит к формированию кристаллического гидрата пентаоксида диванадия (V2O5 ⋅ 0.5H2O, PDF2 27-1366). Увеличение мольного соотношения V2O5 : ДМФА до 1 : 50 в этих же условиях приводит к формированию однофазного триванадата N,N-диметиламмония состава [(CH3)2NH2]V3O7 (рис. 1а). По-видимому, в ходе гидротермальной обработки происходит гидролиз диметилформамида с образованием диметиламина и метановой кислоты, выступающей в качестве восстановителя по отношению к ванадию в степени окисления +5:
(1)
$\begin{gathered} \frac{1}{2}{{{\text{V}}}_{2}}{{{\text{O}}}_{5}} + \frac{1}{3}{{\left( {{\text{C}}{{{\text{H}}}_{3}}} \right)}_{2}}{\text{NCHO}} + \frac{1}{6}{{{\text{H}}}_{2}}{\text{O}}\xrightarrow{\Delta } \\ \to \frac{1}{3}\left[ {{{{\left( {{\text{C}}{{{\text{H}}}_{3}}} \right)}}_{2}}{\text{N}}{{{\text{H}}}_{2}}} \right]{{{\text{V}}}_{3}}{{{\text{O}}}_{7}} + \frac{1}{3}{\text{C}}{{{\text{O}}}_{2}}. \\ \end{gathered} $Рис. 1.
Результаты рентгенофазового (а), термического анализа (б) и ИК-спектроскопии (в) образца [(CH3)2NH2]V3O7, полученного гидротермальной обработкой смеси, содержащей V2O5 и ДМФА в соотношении 1 : 5, при 180°С в течение 7 сут.
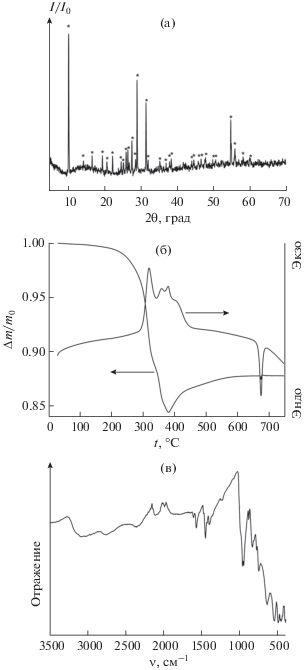
Идентичное соединение в однофазном состоянии было получено в результате гидротермальной обработки реакционных смесей при 180°С в течение 7 сут, а также при 210°С в течение 1 сут, при этом варьирование состава реакционных смесей (в диапазоне мольных соотношений V2O5 : ДМФА от 1 : 5 до 1 : 50) и стартового значения pH (в диапазоне 0.5–3.8) не оказало влияния на фазовый состав продукта синтеза.
По имеющимся литературным данным, фазовый состав продуктов гидротермальной обработки соединений V5+ в присутствии ионов аммония и их производных зачастую очень чувствителен к стартовым значениям pH реакционных смесей [34–36]. Кроме того, анализ продуктов гидротермальной обработки суспензий V2O5 в водных растворах формамида показал, что изменение мольного соотношения реагентов может приводить к получению различных соединений [31].
Соединение [(CH3)2NH2]V3O7 было впервые синтезировано авторами [37] в результате гидротермальной обработки водной суспензии, полученной смешением V2O5, диметиламина, гидроксида лития и уксусной кислоты, при температуре 185°С в течение 4 сут. На основании полученных в [37] структурных данных проведено уточнение параметров элементарной ячейки синтезированного нами соединения, в результате получены следующие значения: a = 7.6598(5), b = 6.6834(3), c = 8.5766(7) Å, β = 98.191(5)° (пр. гр. P21).
Согласно результатам термического анализа (рис. 1б), нагрев триванадата N,N-диметиламмония на воздухе сопровождается разложением этого соединения, протекающим в несколько стадий, по-видимому, связанных с деструкцией органического катиона. Величина потери массы [(CH3)2NH2]V3O7 при нагреве до 380°С составляет ∼15%, что несколько ниже значения, наблюдавшегося ранее (18%) [37]. Такое различие может быть связано с разной скоростью нагрева образцов (10 град/мин в нашем эксперименте и 5 град/мин в эксперименте Лютта и соавт. [37]). В диапазоне температур 380–600°С наблюдается рост массы образца, связанный с процессами окисления соединений ванадия кислородом и формированием V2O5. Общая потеря массы образца [(CH3)2NH2]V3O7 при нагреве от комнатной температуры до 600°С составляет 12.2%, что с высокой точностью соответствует потере массы, рассчитанной для реакции:
В ходе дальнейшего нагрева образца наблюдается выраженный эндотермический эффект при температуре 673°С, связанный с плавлением пентаоксида диванадия [38], что подтверждает схему термических превращений фазы [(CH3)2NH2]V3O7 в ходе нагревания на воздухе.
В ИК-спектре (рис. 1в) фазы [(CH3)2NH2]V3O7 присутствуют полосы, соответствующие валентным колебаниям ν(V=O) (1024–983 см–1) и ν(V–O) (801–767 см–1), деформационным колебаниям δas(V–O) (765–758 см–1) и δs(V–O–V) (535–500 см–1). Указанные полосы наблюдали ранее при анализе оксидов ванадия в степенях окисления +4 и +5, а также их производных [20, 39–42].
Увеличение продолжительности гидротермальной обработки реакционных смесей, содержащих V2O5 и ДМФА, от 1 до 7 сут, а также увеличение температуры гидротермальной обработки от 210 до 250°С при продолжительности синтеза 1 сут приводит к формированию кристаллического диоксида ванадия (VO2(D)), характеризующегося размером частиц до 500 нм, с выходом до 85% (рис. 2). Данные о новой метастабильной D-модификации диоксида ванадия, имеющей моноклинную кристаллическую структуру и изоструктурной вольфрамату(VI) никеля (PDF2 15-0755), были впервые опубликованы Лиу и соавт. в 2011 г. [20]. По имеющимся данным, формирование VO2(D) в гидротермальных условиях протекает через образование промежуточной фазы VO2(B) [43]; в то же время в выбранных нами условиях образование фазы VO2(B) не зафиксировано.
Рис. 2.
Результаты рентгенофазового анализа (а), растровой электронной микроскопии (б) и термического анализа (в) образца VO2(D), полученного гидротермальной обработкой смеси, содержащей V2O5 и ДМФА в соотношении 1 : 5, при 210°С в течение 7 сут. Дифрактограмма (г) порошка, полученного отжигом образца VO2(D) при температуре 300°С в течение 15 мин.
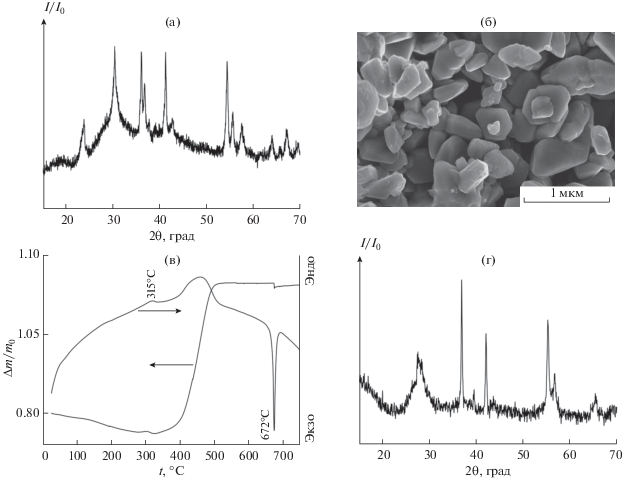
Дополнительно химический состав фазы VO2(D) подтвержден анализом ее термического поведения (рис. 2в, 2г). Результаты термического анализа свидетельствуют о том, что нагрев VO2(D) на воздухе до температур ~300°C приводит к незначительному (~1%) уменьшению массы образца, что может быть связано с удалением физически и химически связанной воды, а также с удалением сорбированных органических соединений, образовавшихся в ходе гидротермального синтеза. При 315°С наблюдается незначительный экзотермический эффект, который, вероятнее всего, соответствует фазовому превращению VO2(D) в VO2(M) [43, 44]. Данное предположение подтверждено результатами РФА порошка, полученного в ходе кратковременной (15 мин) изотермической выдержки (300°С) на воздухе образца фазы VO2(D). В результате такой обработки получен продукт, дифрактограмма которого соответствует VO2(M) (PDF2 43-1051) (рис. 2г). Отметим, что, согласно имеющимся данным, температура фазового превращения VO2(D) в VO2(M) чувствительна к размеру частиц VO2(D) и может находиться в диапазоне от 250 до 400°С [43, 45].
Нагрев образца VO2(D) до 500°С приводит к увеличению его массы, сопровождаемому выраженным и растянутым экзотермическим эффектом, на величину около 10%, что удовлетворительно соответствует изменению массы, рассчитанному по реакции:
Дальнейший нагрев приводит к плавлению образовавшегося V2O5 (эндотермический эффект с минимумом при 672°С).
Гидротермальная обработка реакционных смесей, содержащих V2O5 и ДМФА в мольном соотношении 1 : 5 или 1 : 10, при 250°С в течение 7 сут приводит к формированию порошков однофазного сесквиоксида ванадия со структурой корунда (PDF2 71-0342), характеризующихся субмикронным размером частиц (рис. 3а, 3б). К настоящему времени разработан ряд методов получения высокодисперсных материалов на основе V2O3, при этом большинство из них основано на термическом отжиге оксидных соединений ванадия(IV) или ванадия(V) при температурах выше 450°С в инертной или восстановительной атмосфере [46, 47]. Методы гидротермального синтеза V2O3 развиты в существенно меньшей степени и зачастую требуют использования неустойчивых или дорогостоящих исходных веществ. Так, известны гидротермальные методы получения субмикронных порошков V2O3 со структурой корунда, основанные на восстановлении VO(acac)2 гидразином [48], восстановлении NH4VO3 меркаптоуксусной кислотой [49], восстановлении V2O5 додециламином [50] или 1,6-диаминогексаном [51]. Отметим, что общей особенностью всех перечисленных методов синтеза является высокая температура (200–260°С) и продолжительность (десятки часов) гидротермальной обработки. С точки зрения условий реализации синтеза предложенный нами метод аналогичен уже известным, поскольку он требует проведения гидротермальной обработки при 250°С в течение 7 сут. В то же время он обладает существенным преимуществом с точки зрения доступности исходных веществ (V2O5 и N,N-диметилформамид).
Рис. 3.
Результаты рентгенофазового анализа (а), растровой электронной микроскопии (б) и термического анализа (в) образца V2O3, полученного гидротермальной обработкой смеси, содержащей V2O5 и ДМФА в соотношении 1 : 5, при 250°С в течение 7 сут.

Результаты термического анализа подтверждают формирование V2O3 в выбранных нами условиях (рис. 3в). При нагреве на воздухе выше 400°С наблюдается увеличение массы образца, соответствующее протеканию реакции:
с последующим плавлением образовавшегося пентаоксида диванадия при температуре 673°С.ЗАКЛЮЧЕНИЕ
В настоящей работе впервые проведен гидротермальный синтез ряда оксидных соединений ванадия ([(CH3)2NH2]V3O7, VO2(D) и V2O3) путем восстановления V2O5 N,N-диметилформамидом при температурах 180–250°С. Установлено, что ключевыми параметрами, определяющими фазовый состав продуктов синтеза, являются температура и продолжительность гидротермальной обработки. Предложенный метод синтеза обеспечивает селективное получение перечисленных соединений в однофазном состоянии.
Список литературы
Bahlawane N., Lenoble D. // Chem. Vap. Depos. 2014. V. 20. P. 299. https://doi.org/10.1002/cvde.201400057
Cao Z., Li S., Xie W. et al. // Calphad. 2015. V. 51. P. 241. https://doi.org/10.1016/j.calphad.2015.10.003
Granqvist C.G. // Phys. Scr. 1985. V. 32. P. 401. https://doi.org/10.1088/0031-8949/32/4/026
Kana J.B.K., Ndjaka J.M., Ateba P.O. et al. // Appl. Surf. Sci. 2008. V. 254. P. 3959. https://doi.org/10.1016/j.apsusc.2007.12.021
Bae S.-H., Lee S., Koo H. et al. // Adv. Mater. 2013. V. 25. P. 5098. https://doi.org/10.1002/adma.201302511
Guo Y., Robertson J. // Microelectron. Eng. 2013. V. 109. P. 278. https://doi.org/10.1016/j.mee.2013.03.015
Cabrera R., Merced E., Sepúlveda N. // Phys. Status Solidi A. 2013. V. 210. P. 1704. https://doi.org/10.1002/pssa.201370054
Yang Z., Ko C., Ramanathan S. // Annu. Rev. Mater. Res. 2011. V. 41. P. 337. https://doi.org/10.1146/annurev-matsci-062910-100347
Mjejri I., Rougier A., Gaudon M. // Inorg. Chem. 2017. V. 56. P. 1734. https://doi.org/10.1021/acs.inorgchem.6b02880
Shvets P., Dikaya O., Maksimova K., Goikhman A. // J. Raman Spectrosc. 2019. V. 50. P. 1226. https://doi.org/10.1002/jrs.5616
Parker J.C., Geiser U.W., Lam D.J. et al. // J. Am. Ceram. Soc. 1990. V. 73. P. 3206. https://doi.org/10.1111/j.1151-2916.1990.tb06438.x
Wu C., Feng F., Xie Y. // Chem. Soc. Rev. 2013. V. 42. P. 5157. https://doi.org/10.1039/c3cs35508j
Zhang Z., Gao Y., Chen Z. et al. // Langmuir. 2010. V. 26. P. 10738. https://doi.org/10.1021/la100515k
Morin F.J. // Phys. Rev. Lett. 1959. V. 3. P. 34. https://doi.org/10.1103/PhysRevLett.3.34
Berezina O.Y., Velichko A.A., Lugovskaya L.A. et al. // Inorg. Mater. 2007. V. 43. P. 505. https://doi.org/10.1134/S0020168507050123
Schwingenschlögl U., Eyert V. // Ann. der Phys. 2004. V. 13. P. 475. https://doi.org/10.1002/andp.200410099
Oka Y., Yao T., Yamamoto N. // J. Solid State Chem. 1990. V. 86. P. 116. https://doi.org/10.1016/0022-4596(90)90121-D
Théobald F., Cabala R., Bernard J. // J. Solid State Chem. 1976. V. 17. P. 431. https://doi.org/10.1016/S0022-4596(76)80013-8
Meyer L.M., Haushalter R.C., Hagrman D. et al. // J. Solid State Chem. 2002. V. 138. P. 178. https://doi.org/10.1006/jssc.1997.7575
Liu L., Cao F., Yao T. et al. // New J. Chem. 2012. V. 36. P. 619. https://doi.org/10.1039/c1nj20798a
Wu C., Hu Z., Wang W. et al. // Chem. Commun. 2008. P. 3891. https://doi.org/10.1039/b806009f
Qazilbash M.M., Schafgans A.A., Burch K.S. et al. // Phys. Rev. B. 2008. V. 77. P. 115121. https://doi.org/10.1103/PhysRevB.77.115121
Andreev V.N., Klimov V.A., Kompan M.E. // Phys. Solid State. 2017. V. 59. P. 2441. https://doi.org/10.1134/S1063783417120046
Wessel C., Reimann C., Müller A. et al. // J. Comput. Chem. 2012. V. 33. P. 2102. https://doi.org/10.1002/jcc.23046
Weber D., Stork A., Nakhal S. et al. // Inorg. Chem. 2011. V. 50. P. 6762. https://doi.org/10.1021/ic200799n
Bergerud A., Selbach S.M., Milliron D.J. // ACS Nano. 2016. V. 10. P. 6147. https://doi.org/10.1021/acsnano.6b02093
Reimann C., Weber D., Lerch M., Bredow T. // J. Phys. Chem. C. 2013. V. 117. P. 20164. https://doi.org/10.1021/jp406622u
Li M., Magdassi S., Gao Y., Long Y. // Small. 2017. V. 13. P. 1701147. https://doi.org/10.1002/smll.201701147
Li W., Ji S., Li Y. et al. // RSC Adv. 2014. V. 4. P. 13026. https://doi.org/10.1039/C3RA47666A
Park H.-K., Kim G. // Solid State Ionics. 2010. V. 181. P. 311. https://doi.org/10.1016/j.ssi.2010.01.011
Teplonogova M.A., Yapryntsev A.D., Baranchikov A.E., Ivanov V.K. // Transit. Met. Chem. 2019. V. 44. P. 25. https://doi.org/10.1007/s11243-018-0265-x
Bates R.G., Coetzee J.F., Bishop E. et al. // Pure Appl. Chem. 1977. V. 49. P. 885. https://doi.org/10.1351/pac197749060885
Petříček V., Dušek M., Palatinus L. // Z. Krist. Cryst. Mater. 2014. V. 229. P. 345. https://doi.org/10.1515/zkri-2014-1737
Chirayil T., Zavalij P.Y., Whittingham M.S. // Chem. Mater. 1998. V. 10. P. 2629. https://doi.org/10.1021/cm980242m
Zakharova G.S., Baklanova I.V., Suntsov A.Y. et al. // Russ. J. Inorg. Chem. 2016. V. 61. P. 1584. https://doi.org/10.1134/S0036023616120214
Podval’naya N.V., Zakharova G.S., Slepukhin P.A. // Russ. J. Inorg. Chem. 2019. V. 64. P. 994. https://doi.org/10.1134/S0036023619080102
Lutta S.T., Chernova N.A., Zavalij P.Y., Whittingham M.S. // J. Mater. Chem. 2004. V. 14. P. 2922. https://doi.org/10.1039/b405150e
Jognston C.T. CRC Handbook of Chemistry and Physics, 94th Edition / Eds. Haynes W.M., Lide D.R., Bruno T.J. New York: CRC Press LLC, 2013.
Zakharova G.S., Volkov V.L. // Russ. Chem. Rev. 2003. V. 72. P. 311. https://doi.org/10.1070/RC2003v072n04ABEH000762
Suvorova O.N., Wöhrle D., Shupak E.A. et al. // Macroheterocycles. 2010. V. 3. P. 134. https://doi.org/10.6060/mhc2010.2-3.134
Vandenborre M.T., Prost R., Huard E., Livage J. // Mater. Res. Bull. 1983. V. 18. P. 1133. https://doi.org/10.1016/0025-5408(83)90157-5
Repelin Y., Husson E., Abello L., Lucazeau G. // Spectrochim. Acta A. 1985. V. 41. P. 993. https://doi.org/10.1016/0584-8539(85)80063-5
Song Z., Zhang L., Xia F. et al. // Inorg. Chem. Front. 2016. V. 3. P. 1035. https://doi.org/10.1039/c6qi00102e
Teixeira D., Quesada-Cabrera R., Powell M.J. et al. // New J. Chem. 2017. V. 41. P. 9216. https://doi.org/10.1039/c7nj02165h
Luo Y., Li M., Li G. et al. // CrystEngComm. 2015. V. 17. P. 5614. https://doi.org/10.1039/c5ce00873e
Santulli A.C., Xu W., Parise J.B. et al. // Phys. Chem. Chem. Phys. 2009. V. 11. P. 3718. https://doi.org/10.1039/b822902c
Corr S.A., Grossman M., Shi Y. et al. // J. Mater. Chem. 2009. V. 19. P. 4362. https://doi.org/10.1039/b900982e
Bai Y., Jin P., Ji S. et al. // Ceram. Int. 2013. V. 39. P. 7803. https://doi.org/10.1016/j.ceramint.2013.03.040
Ji H., Liu D., Cheng H. et al. // Mater. Sci. Eng. B. 2017. V. 217. P. 1. https://doi.org/10.1016/j.mseb.2017.01.003
Sarkar A., Sinha A.K., Mitra S. // Electrochim. Acta. 2019. V. 299. P. 914. https://doi.org/10.1016/j.electacta.2019.01.076
Sediri F., Gharbi N. // Mater. Sci. Eng. B. 2005. V. 123. P. 136. https://doi.org/10.1016/j.mseb.2005.07.010
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии