

Журнал неорганической химии, 2020, T. 65, № 4, стр. 517-521
Влияние компонентного состава раствора EtOH : H2O на устойчивость глицилглицинатных комплексов кобальта(II)
В. А. Исаева a, А. С. Молчанов b, *, М. В. Шишкин b, К. А. Кипятков a, В. А. Шарнин a
a Ивановский государственный химико-технологический университет
153000 Иваново, Шереметевский пр-т, 7, Россия
b Костромской государственный университет
156005 Кострома, ул. Дзержинского, 17, Россия
* E-mail: mas_07@inbox.ru
Поступила в редакцию 28.10.2019
После доработки 13.11.2019
Принята к публикации 27.11.2019
Аннотация
Константы устойчивости глицилглицинатных комплексов кобальта(II) в растворителе вода–этанол переменного состава определены потенциометрическим методом при температуре 298 K и ионной силе растворов 0.1 (NaClO4). Установлено, что с ростом содержания этанола в растворе устойчивость комплексов кобальта(II) с глицилглицинат-ионом возрастает. Полученные значения констант устойчивости глицилглицината кобальта(II) сопоставлены с литературными данными по устойчивости комплексов никеля(II) и меди(II) с анионом глицилглицина. Дана оценка вкладов пересольватации реагентов в изменение энергии Гиббса реакции образования глицилглицината кобальта(II) в растворителе вода–этанол. Показано, что увеличение устойчивости комплексов с глицилглицинат-ионом обусловлено в основном ослаблением сольватации лиганда при переходе от воды к водно-этанольным растворителям.
ВВЕДЕНИЕ
Изучение влияния состава растворителя на процессы комплексообразования является важным направлением исследований физической химии растворов. Это обусловлено значимостью координационных равновесий в водных и неводных растворителях, а также представляет теоретический интерес для установления взаимосвязи процессов сольватации и комплексообразования. К настоящему времени накоплен достаточно большой объем термодинамических данных о реакциях комплексообразования ионов d-металлов с лигандами аминного и карбоксилатного типа в водно-органических растворителях и дана количественная характеристика сольватационных вкладов реагентов в термодинамику реакций комплексообразования [1]. Поведение полифункциональных соединений в неводных растворителях имеет ряд особенностей, которые к настоящему времени недостаточно изучены.
Глицилглицин  представляет собой дипептид, построенный из двух молекул аминокислоты глицина посредством
пептидной связи. С большинством d-металлов анион глицилглицина образует комплексы за счет взаимодействия атома азота
аминогруппы и атома кислорода пептидной группы [2].
представляет собой дипептид, построенный из двух молекул аминокислоты глицина посредством
пептидной связи. С большинством d-металлов анион глицилглицина образует комплексы за счет взаимодействия атома азота
аминогруппы и атома кислорода пептидной группы [2].
Координационные соединения аниона глицил-глицина с ионами микроэлементов играют значительную роль в биологических процессах, протекающих в организмах [3]. Изучение устойчивости комплексов d-металлов c биолигандами в смешанных растворителях необходимо для подбора оптимальных условий синтеза координационных соединений [4, 5]. Ранее изучена устойчивость глицилглицинатных комплексов меди(II) и никеля(II) в различных водно-органических растворителях [6–9]. В продолжение этих исследований в настоящей работе изучено влияние растворителя вода–этанол переменного состава на устойчивость комплексов глицилглицинат-иона с ионом кобальта(II).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Константы равновесия реакций комплексообразования кобальта(II) с глицилглицинат-ионом определены методом потенциометрического титрования с использованием стеклянного электрода, который широко применяется для исследования равновесных процессов в водно-этанольных смесях [10], и хлорсеребряного электрода, внутренний раствор которого приготовлен на основе водно-этанольного растворителя соответствующего состава с целью уменьшения диффузионного потенциала на концах электролитического мостика. Дозировку титранта осуществляли весовым способом с помощью микрошприца с последующим пересчетом на объем. Общий объем титранта составлял ∼3.5 мл, объем одной порции – ∼0.2 мл. Титрантом являлся раствор глицилглицината натрия (8 × 10–1 моль/л). В ячейке находился водно-этанольный раствор (50 мл), содержащий Co(ClO4)2 (2 × 10–2 моль/л) и HClO4 (1 × × 10–2 моль/л). Титрование проводили до значения рН не более 7.5. Измерения выполняли при температуре 298 K и ионной силе µ = 0.1 M на фоне перхлората натрия.
В работе использовали гексагидрат перхлората кобальта фирмы “Sigma-Aldrich”. Содержание основного вещества – Co(ClO4)2 · 6H2O – уточняли методом прямого комплексонометрического титрования этилендиаминтетраацетатом натрия (ЭДТА) с индикатором мурексидом в присутствии аммиачного буфера при рН ~ 6. Перхлорат натрия (ч.) очищали перекристаллизацией из водного раствора. Раствор глицилглицината натрия готовили по точным навескам эквимолярных количеств глицилглицина (Sigma-Aldrich, содержание основного вещества ≥99%) и бескарбонатного насыщенного раствора NaOH. Гидроксид натрия имел квалификацию “х. ч.”. Этанол (EtOH) марки “ректификат” перегоняли, остаточное содержание воды учитывали при приготовлении растворов. Для предотвращения окисления Co2+/Co3+ воду (бидистиллят), используемую для приготовления растворов, дополнительно деаэрировали с целью удаления растворенного кислорода. Титрование проводили в среде инертного газа.
Расчет констант устойчивости комплексов по результатам потенциометрического титрования проводили по программе PHMETR [11]. При вычислении констант устойчивости комплексов в программе использовали алгоритм итеративного поиска при заданных начальных приближениях констант минимума целевой функции:
(1)
$F = \mathop \sum \limits_{i = 1}^n {{\left( {{\text{p}}{{{\text{H}}}_{{{\text{расч}}}}} - {\text{p}}{{{\text{H}}}_{{{\text{эксп}}}}}} \right)}^{2}}{{{\omega }}_{i}},$Погрешность численных значений констант оценивали на основе статистической обработки результатов 3–4 параллельных опытов в каждой точке составов растворителя.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
В водном растворе ион двухвалентного кобальта образует с анионом глицилглицина (GG¯) моно- и бис-координированные комплексы [2]:
(2)
${\text{C}}{{{\text{о}}}^{{{\text{2}} + }}} + {\text{G}}{{{\text{G}}}^{ - }} \rightleftarrows {\text{ }}{{[{\text{CоGG}}]}^{ + }},~~~{\text{lg}}{{K}_{{\text{1}}}},$(3)
${{[{\text{CоGG}}]}^{ + }} + {\text{G}}{{{\text{G}}}^{ - }} \rightleftarrows {\text{ }}[{\text{CоGG}}]{{,}_{{\text{ }}}}~~{\text{lg}}{{K}_{{\text{2}}}}.$При обработке данных потенциометрического титрования в расчетной схеме программы PHMETR [11] были учтены реакции образования моно- и бис-глицилглицинатов кобальта(II), а также процессы кислотно-основных взаимодействий глицилглицина в водно-этанольных растворах и автопротолиза водно-этанольного растворителя переменного состава, константы равновесия которых приведены соответственно в работах [7] и [12]. Образование комплекса кобальта(II) с глицилглицинат-ионом состава 1 : 3 расчетами не подтверждалось ни в водном, ни в водно-этанольном растворе. Проверочные расчеты свидетельствовали об отсутствии процесса гидролиза кобальта(II), поскольку введение в расчетную схему реакции образования гидроксокомплекса кобальта(II), константа равновесия которой для водного раствора была взята из [13], не изменяло значение критериальной функции программы PHMETR [11] и не влияло на значения рассчитываемых констант lgK1 и lgK2. Полученные нами значения констант устойчивости моно- и бис-глицилглицинатных комплексов кобальта(II) для водного раствора находятся в хорошем соответствии с литературными данными (табл. 1). Найденное значение константы устойчивости монолигандного комплекса кобальта(II) lg K1 = 3.72 при содержании этанола в растворе 0.1 мол. д. сопоставимо с литературным значением lg K1 = 3.6 [18], полученным при ХEtOH = 0.09 мол. д., Т = 298 K, µ = 0.1 (KNO3).
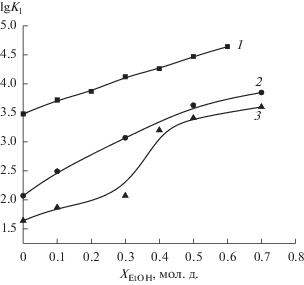
Значения констант устойчивости глицилглицинатов кобальта(II), определенные в водно-этанольных растворах с различной концентрацией этанола (табл. 2), показывают, что повышение содержания этанола в растворе способствует увеличению устойчивости образующихся комплексов. Аналогичное влияние водно-этанольного растворителя наблюдалось при образовании комплексов кобальта(II) с анионами янтарной [19] и малеиновой кислот [20] (рис. 1).
Таблица 2.
Константы устойчивости глицилглицинатных комплексов кобальта(II) в водно-этанольных растворах переменного состава, Т = 298 K, µ = 0.1 (NaClO4)
| Параметр | Мольная доля этанола | ||||||
|---|---|---|---|---|---|---|---|
| 0.0 | 0.1 | 0.2 | 0.3 | 0.4 | 0.5 | 0.6 | |
| lg K1 ± 0.04 | 3.48 | 3.72 | 3.87 | 4.12 | 4.26 | 4.47 | 4.64 |
| lg K2 ± 0.07 | 2.66 | 2.88 | 2.94 | 3.11 | 3.24 | 3.36 | 3.59 |
Сравнение экспериментальных данных с константами устойчивости глицилглицинатных комплексов никеля(II) [7] и меди(II) [6] (рис. 2) показало, что для всех составов водно-этанольного растворителя соотношение констант устойчивости комплексов с ионами двухвалентных металлов соответствует ряду Ирвинга–Уильямса (Co2+ < Ni2+ < Cu2+).
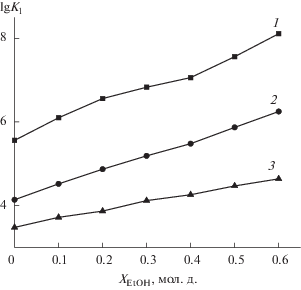
Прирост констант устойчивости моноглицил-глицинатных комплексов никеля(II) [7] и меди(II) [6] в водно-этанольных смесях составляет более двух логарифмических единиц (при ХEtOH = 0.6), что превышает изменение устойчивости глицил-глицината кобальта(II) в данном растворителе (рис. 2). Однако изменение сольватного состояния иона-комплексообразователя в смешанном растворителе не вносит определяющий вклад в изменение устойчивости образующихся комплексов, поскольку при соизмеримом приросте lg K1 глицилглицината никеля(II) и меди(II) в водно-этанольном растворе изменение энергии Гиббса пересольватации ионов этих металлов различно. Перенос иона никеля(II) из воды в водно-этанольный растворитель [21] характеризуется отрицательным (при XEtOH > 0.15 мол. д.) значением энергии Гиббса пересольватации, в то время как изменение энергии Гиббса пересольватации иона меди(II) положительно во всей области составов водно-этанольного растворителя [22].
Для оценки вклада пересольватации лиганда в смешанном растворителе в изменение энергии Гиббса реакции комплексообразования рассчитали значения ΔG° переноса из воды в водно-этанольные смеси глицилглицинат-иона (GG¯) по уравнению:
(4)
$\begin{gathered} {{\Delta }_{{tr}}}G_{r}^{\circ } = {{\Delta }_{{tr}}}G^\circ ({\text{GG}}) + \\ + {{\Delta }_{{tr}}}G^\circ ({{H}^{ + }}){\text{ }} - {{\Delta }_{{tr}}}G^\circ ({\text{HG}}{{{\text{G}}}^{ \pm }}). \\ \end{gathered} $Для этого использовали литературные данные об изменении энергии Гиббса реакции диссоциации глицилглицина (ΔtrGr) [7] и ΔtrG° протона [23] в водно-этанольном растворителе переменного состава. Значения изменения энергии Гиббса переноса глицилглицина (HGG±) из воды в растворитель вода–этанол переменного состава были рассчитаны по уравнению:
(5)
${{\Delta }_{{tr}}}G^\circ \left( {{\text{HG}}{{{\text{G}}}^{ \pm }}} \right) = - 2.303RT{\text{lg}}\left( {{{C}_{{mix}}}/{{C}_{{\text{w}}}}} \right),$Численные значения растворимости глицил-глицина (мг/мл) при 298 K приведены в работе [24] в диапазоне составов водно-этанольного растворителя 0–60 об. %. Данные [24] пересчитаны в молярную концентрацию (моль/л) и приведены к шкале содержания этанола в растворе, выраженного в мольных долях через плотность водно-этанольных смесей при 298 K [25].
Из-за отсутствия данных об изменении сольватного состояния иона кобальта(II) в растворителе вода–этанол по полученным значениям ΔtrG° глицилглицинат-иона и ΔtrG реакции образования [CоGG]+ рассчитали величину различий в изменении энергии Гиббса пересольватации комплексной частицы и Со2+ в водно-этанольном растворителе:
(6)
$\begin{gathered} {{\Delta }_{{tr}}}G_{{r1}}^{^\circ } = {{\Delta }_{{tr}}}G^\circ ({{[{\text{CоGG}}]}^{ + }}) - \\ - \,\,{{\Delta }_{{tr}}}G^\circ \left( {{\text{С}}{{{\text{о}}}^{{2 + }}}} \right) - {{\Delta }_{{tr}}}G^\circ ({\text{GG}}). \\ \end{gathered} $В расчетах по уравнениям (4) и (6) значения ΔtrG реакций диссоциации глицилглицина и образования глицилглицината кобальта(II), полученные при µ = 0.1 M, принимали за стандартные величины.
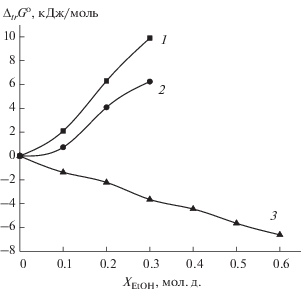
Как показывает рис. 3, уменьшение энергии Гиббса реакции образования [CоGG]+ в основном определяется ослаблением сольватации глицилглицинат-иона в водно-этанольных смесях при частичной компенсации этого вклада различием в изменении сольватного состояния комплексного и центрального ионов. Подобное соотношение сольватационных вкладов реагентов в изменение энергии Гиббса реакции в водно-органических растворах установлено для большинства процессов комплексообразования d-металлов с N-, O-донорными лигандами [1].
Рис. 3.
Влияние состава водно-этанольного растворителя на изменение энергии Гиббса реакции образования глицилглицината кобальта(II) и пересольватации реагентов: 1 – ΔtrG°(GG¯), 2 – ΔtrG°([СоGG]+ – Co2+), 3 – Δtr$G_{{r1}}^{^\circ }$.
В работе [1] показано, что термодинамические характеристики реакции комплексообразования находятся в пределах изменения соответствующих характеристик лиганда (L):
Коэффициенты различий (αdif) для комплексов ионов d-металлов с N-, O-донорными лигандами в большинстве случаев равны 0.6–0.8. Рассчитанный для процесса образования [CоGG]+ коэффициент различий при концентрации этанола 0.3 мол. д. составил 0.63.
Таким образом, при изучении процесса образования глицилглицината кобальта(II) общие закономерности изменения термодинамических параметров реакций комплексообразования и пересольватации лигандов в водно-органических растворителях, установленные для аминных и карбоксилатных комплексов d-металлов [1], нашли подтверждение.
Список литературы
Шарнин В.А. // Изв. вузов. Химия и хим. технология. 2005. Т. 48. Вып. 7. С. 44.
Rabin B.R. // Trans. Faraday Soc. 1956. V. 52. P. 1130.
Комов В.П., Шведова В.Н. Биохимия: учеб. для вузов. М.: Дрофа, 2004. 638 с.
Malinina E.A., Kochneva I.K., Avdeeva V.V. et al. // Russ. J. Inorg. Chem. 2019. V. 64. № 10. P. 1210. [Малинина Е.А., Кочнева И.К., Авдеева В.В. и др. // Журн. неорган. химии. 2019. Т. 64. № 10. С. 1031.]https://doi.org/10.1134/S0044457X19100088
Sharutin V.V., Sharutinf O.K., Senchurin V.S. // Russ. J. Inorg. Chem. 2019. V. 64. № 8. P. 1025. [Шарутин В.В., Шарутина О.К., Сенчурин В.С. // Журн. неорган. химии. 2019. Т. 64. № 8. С. 862.]https://doi.org/10.1134/S0044457X19080130
Isaeva V.A., Molchanov A.S., Kipyatkov K.A. et al. // Russ. J. Phys. Chem. 2019. V. 93. № 8. P. 1460. [Исаева В.А., Молчанов А.С., Кипятков К.А. и др. // Журн. физ. химии. 2019. Т. 93. № 8. С. 1164.]https://doi.org/10.1134/S0036024419080107
Naumov V.V., Isaeva V.A., Sharnin V.A. // Russ. J. Inorg. Chem. 2011. V. 56. № 7. P. 1139. [Наумов В.В., Исаева В.А., Шарнин В.А. // Журн. неорган. химии. 2011. Т. 56. № 7. С. 1208.]
Naumov V.V., Isaeva V.A., Kovaleva Yu. A., Sharnin V.A. // Russ. J. Phys. Chem. 2013. V. 87. № 7. P. 1135. [Наумов В.В. Исаева В.А., Ковалева Ю.А., Шарнин В.А. // Журн. физ. химии. 2013. Т. 87. № 7. С. 1160.]https://doi.org/10.7868/S0044453713070236
Isaeva V.A., Naumov V.V., Sharnin V.A. // Russ. J. Coord. Chem. 2009. V. 35. № 11. P. 868. [Исаева В.А., Наумов В.В., Шарнин В.А. // Коорд. химия. 2009. Т. 35. № 11. С. 878.]https://doi.org/10.1134/S107032840911013X
Батлер Дж. // Электрохимия металлов в неводных растворах / Под ред. Колотыркина Я.М. М.: Мир, 1977. 440 с.
Бородин В.А., Козловский Е.В., Васильев В.П. // Журн. неорган. химии. 1986. Т. 31. № 1. С. 10.
Woollej E.H., Hurkot D.G., Herber L.G. // J. Phys. Chem. 1970. V. 74. № 22. P. 3908.
Bolzan J.A., Arvia A.J. // Electrochim. Acta. 1962. V. 7. P. 589.
Palade D.M., Gannova Yu.N. // Russ. J. Coord. Chem. 2003. V. 29. № 2. P. 106. [Паладе Д.М., Ганнова Ю.Н. // Коорд. химия. 2003. Т. 29. № 2. С. 113.]
Harris W.R., Martell A.E. // J. Am. Chem. Soc. 1977. V. 99. P. 6746.
Biester J., Ruoff P. // J. Am. Chem. Soc. 1959. V. 81. P. 6517.
Петров H.B., Набоков В.С., Жаданов Б.В. и др. // Журн. физ. химии. 1976. Т. 50. № 9. С. 2208.
Boraei A., Ahmed I. // Synth. React. Inorg. Met.-Org. Chem. 2002. V. 32. P. 981.
Tukumova N.V., Tran Thi Dieu Thuan, Usacheva T.R. et al. // Russ. J. Phys. Chem. 2017. V. 91. № 4. P. 662. [Тукумова Н.В., Тхуан Чан Тхи Зъеу, Усачева Т.Р. и др. // Журн. физ. химии. 2017. Т. 91. № 4. С. 639.]https://doi.org/10.7868/S0044453717040318
Тукумова Н.В., Усачева Т.Р., Чан Тхуан, Шарнин В.А. // Изв. вузов. Химия и хим. технология. 2012. Т. 55. Вып. 9. С. 16.
Невский А.В., Шарнин В.А., Шорманов В.А., Крестов Г.А. // Коорд. химия. 1983. Т. 9. № 3. С. 391.
Lewandowski A. // Electrochim. Acta. 1984. V. 29. P. 547.
Kalidas C., Hefter G., Marcus Y. // Chem. Rev. 2000. V. 100. № 3. P. 819.
Lu J., Wang X.-J., Yang X., Ching C.-B. // J. Chem. Eng. Data. 2006. V. 51. P. 1593.
Yilmaz H. // Turk. J. Phys. 2002. V. 26. P. 243.
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии