

Журнал неорганической химии, 2020, T. 65, № 3, стр. 296-301
Влияние монокристаллической подложки r-Al2O3 на процесс роста частиц Ti1 – xVxO2 в условиях гидротермального синтеза
О. Н. Макаревич a, b, А. В. Иванов a, b, А. И. Гаврилов a, А. М. Макаревич a, b, О. В. Бойцова a, b, *
a Институт общей и неорганической химии им. Н.С. Курнакова РАН
119991 Москва, Ленинский пр-т, 31, Россия
b Московский государственный университет имени М.В. Ломоносова
119991 Москва, Ленинские горы, 1, Россия
* E-mail: boytsova@gmail.com
Поступила в редакцию 15.10.2019
После доработки 27.10.2019
Принята к публикации 01.11.2019
Аннотация
Показана возможность формирования методом гидротермального синтеза текстурированных покрытий Ti1– xVxO2 на поверхности монокристаллических подложек r-Al2O3 с использованием в качестве прекурсоров гексафтортитаната аммония и оксалатного комплекса ванадила. Полученные образцы охарактеризованы методами рентгеновской дифракции, растровой электронной микроскопии и рентгеноспектрального микроанализа.
ВВЕДЕНИЕ
Традиционно метод гидротермального синтеза используется для получения материалов, где требуется смягчение условий синтеза: уменьшение времени и температуры протекания реакций, компромиссные решения для совместного соосаждения, невозможного или ресурсозатратного в сухих способах синтеза [1]. Особенно актуально развитие этого направления при получении метастабильных соединений, в том числе покрытий с новыми свойствами или стабилизированных фаз, неустойчивых в объемном виде [2].
Одним из таких объектов являются твердые растворы пленок диоксида титана-ванадия, имеющие широкое применение в виде наночастиц [3, 4] или тонких пленок [5, 6]. В литературе информация о способах получения таких твердых растворов появилась не так давно [7–9]. В большинстве работ используется метод химического осаждения из газовой фазы [10], золь-гель метод [11], соосаждение [11–13] и физические методы [3, 14, 15].
Диоксид ванадия как в виде порошка, так и в виде пленок редко может быть получен в однофазном состоянии. Это связано с узкой областью кислородной стехиометрии и сосуществованием с фазами Магнели VnO2n – 1 и Вадсли VmO2m+ 1 [16]. В статье предложен новый способ получения покрытий твердого раствора Ti1 – xVxO2, стабилизированного за счет образующейся структуры анатаза в присутствии устойчивого фторсодержащего прекурсора, используемого для формирования высокоструктурированных объектов на основе диоксида титана [17].
В рамках данной работы впервые продемонстрирована возможность получения текстурированных покрытий смешанного состава Ti1– xVxO2 методом гидротермального синтеза на монокристаллических подложках r-Al2O3, которые широко используются для получения эпитаксиальных пленок TiO2 [18] и VO2 [19].
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В качестве исходных соединений использовали гексафтортитанат аммония (ос. ч., Aldrich), изопропиловый спирт и оксалатный комплекс ванадила (NH4)2[(VO)2(C2O4)3(H2O)2]. Оксалатный комплекс ванадила синтезировали по методике, описанной в работе [20]. Состав комплекса подтвержден методами термогравиметрического анализа (ТГА) и ИК-спектроскопии. По результатам ТГА для стадии дегидратации рассчитано: 8%, найдено: 9%. Для следующей стадии образования V2O5 из безводного комплекса рассчитано: 53%, найдено: 52%. В ИК-спектре присутствуют следующие характеристические частоты: 3050–3200 ν(OH), 1594 νas(COO–), 1398 νs(COO–) + + δ(N${\text{H}}_{4}^{ + }$), 1287 ν(C–O), 974 ν(V=O), 804 см–1 δ(V–O).
Синтез пленок и порошков Ti1 – xVxO2 осуществляли при совместной гидротермальной обработке титансодержащего прекурсора (NH4)2TiF6 и прекурсора ванадия (NH4)2[(VO)2(C2O4)3(H2O)2]. Мольное соотношение металлов в исходной смеси составляло Ti : V = 2 : 1.
В раствор (NH4)2[(VO)2(C2O4)3(H2O)2] определенной концентрации (0.02, 0.05, 0.07 и 0.1 моль/л) добавляли рассчитанное количество порошка гексафтортитаната аммония и перемешивали при комнатной температуре до полного растворения компонентов. Полученный раствор переливали в тефлоновый вкладыш объемом 10 мл до степени заполнения 90% и помещали в него монокристаллическую подложку из r-Al2O3. Тефлоновый вкладыш размещали в стальном автоклаве и проводили синтез при 195°С в течение 6 ч. В результате гидротермального синтеза образовался бирюзовый раствор с темным осадком и пленкой темного цвета на подложках. Пленку и твердый осадок промывали несколько раз в дистиллированной воде, спирте и оставляли на воздухе до полного высыхания.
Термический отжиг образцов порошков и пленок осуществляли в атмосфере аргона при 400°С в течение 4 ч.
Рентгенофазовый анализ (РФА) порошковых образцов проводили на дифрактометре Bruker D8 Advance (CuKα-излучение) в диапазоне углов 2θ 10°–60° при скорости вращения гониометра 3 град/мин, пленок – на дифрактометре Smart LAB. Индицирование дифрактограмм выполняли с использованием базы данных PDF2 (2012).
Растровую электронную микроскопию (РЭМ) и рентгеноспектральный микроанализ полученных образцов проводили с использованием микроскопа Carl Zeiss NVision 40, оснащенного анализатором Oxford Instruments X-Max (80 мм2).
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Гидротермальный синтез
Для реализации процесса соосаждения оксидных фрагментов V(+4) и Ti(+4) в твердый раствор использовали два подобранных прекурсора, которые представляли собой координационные соединения с хорошей растворимостью в воде и высокой устойчивостью к гидролизу при обычном давлении. Превращение указанных комплексов в оксидные соединения происходит в гидротермальных условиях. Реакции разложения прекурсоров можно представить следующим образом:
(1)
${{{\text{(N}}{{{\text{H}}}_{{\text{4}}}}{\text{)}}}_{{\text{2}}}}{\text{Ti}}{{{\text{F}}}_{{\text{6}}}} + 2{{{\text{H}}}_{{\text{2}}}}{\text{O}} = {\text{Ti}}{{{\text{O}}}_{2}} + 2{\text{N}}{{{\text{H}}}_{{\text{4}}}}{\text{F}} + 2{\text{HF}},$(2)
$\begin{gathered} {{\left( {{\text{N}}{{{\text{H}}}_{{\text{4}}}}} \right)}_{{\text{2}}}}\left[ {{{{\left( {{\text{VO}}} \right)}}_{{\text{2}}}}{{{\left( {{{{\text{C}}}_{{\text{2}}}}{{{\text{O}}}_{{\text{4}}}}} \right)}}_{{\text{3}}}}{{{\left( {{{{\text{H}}}_{{\text{2}}}}{\text{O}}} \right)}}_{{\text{2}}}}} \right] = \\ = 2{\text{V}}{{{\text{O}}}_{2}} + 3{\text{CO}} + 2{\text{C}}{{{\text{O}}}_{2}} + {{\left( {{\text{N}}{{{\text{H}}}_{4}}} \right)}_{2}}{\kern 1pt} {\text{C}}{{{\text{O}}}_{3}} + 2{{{\text{H}}}_{{\text{2}}}}{\text{O}}. \\ \end{gathered} $Целью данной работы является совместное разложение прекурсоров титана и ванадия для протекания реакции образования твердого раствора:
(3)
$\left( {1{\text{ }}--x} \right){\text{Ti}}{{{\text{O}}}_{2}} + x{\text{V}}{{{\text{O}}}_{2}} = {\text{T}}{{{\text{i}}}_{{1--{\text{ }}x}}}{{{\text{V}}}_{x}}{{{\text{O}}}_{2}}.$В табл. 1 приведены условия получения пленок и порошков в ходе экспериментов. Порошок в количестве, необходимом для проведения РФА, удалось получить только в эксперименте 1 с максимально высокой концентрацией прекурсоров.
Таблица 1.
Условия получения образцов методом гидротермального синтеза
| Параметр | Номер эксперимента | |||
|---|---|---|---|---|
| 1 | 2 | 3 | 4 | |
| CV, моль/л | 0.1 | 0.07 | 0.05 | 0.02 |
| CTi, моль/л | 0.2 | 0.14 | 0.1 | 0.04 |
| Порошок | + | – | – | – |
| Пленка | + | + | + | + |
На рис. 1 представлена рентгенограмма порошка, из которой видно, что расположение линий близко к рефлексам фазы анатаза TiO2(А). Заметен некоторый сдвиг линий относительно сигналов фазы анатаза, что может быть связано с вхождением ванадия в структуру диоксида титана. Индицирование рентгенограммы порошка позволило получить следующие результаты: а = 3.80, с = = 9.48 Å, что отличается от структуры TiO2(A): a = = 3.78, c = 9.51 Å.
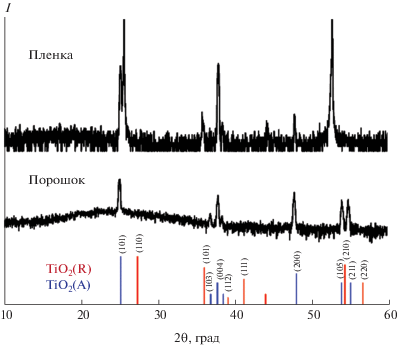
На рентгенограмме пленки, полученной в тех же условиях (концентрация прекурсора ванадия 0.1 моль/л), можно отметить образование текстуры в пленочных образцах, о чем свидетельствуют выделяющиеся по интенсивности рефлексов линии на рентгенограмме пленочного образца. Направление текстуры определено как (004) и (101) анатаза, что отвечает наиболее быстро растущим граням кристаллов диоксида титана [21]. На рентгенограмме пленки есть дополнительный сигнал 36°, который отсутствует на рентгенограмме порошка. Рефлекс следует отнести к рефлексу рутила (101) TiO2(R), который проявляется на пленочных образцах TiO2/r-Al2O3, полученных газофазными методами [18], и указывает на эффект эпитаксиальной стабилизации высокотемпературной рутильной фазы диоксида титана в условиях гидротермального синтеза. Данное явление весьма интересно, так как эпитаксиальный рост происходит исключительно при низких температурах – всего 195°С.
На рис. 2 приведены микрофотографии пленки и порошка, полученных в аналогичных условиях. Пленка на подложке образована сериями микрочастиц различных размеров: ∼100 нм, 1 мкм и слипшихся частиц с размером грани 4–5 мкм в форме, близкой к квадратной. Порошок образован чешуйчатыми частицами, каждая из которых состоит из микрочастиц размером 1–5 мкм. Микроморфология порошка и составляющих частиц покрытия носит родственный характер.

По данным микроанализа были определены соотношения Ti : V, в порошке и пленках они равны соответственно 5.30(5) : 1 и 4.90(5) : 1. Отличие соотношения Ti : V от стехиометрического объясняется разницей в кинетике разложения использованных прекурсоров титана и ванадия. Пленки по сравнению с порошком содержат меньше титана, возможно, вследствие более высокой склонности соединений титана к гидролизу и обеднению раствора вследствие выделения титансодержащего осадка.
Влияние концентрации раствора
Для выявления влияния концентрации исходного раствора ванадиевого прекурсора на морфологию и состав полученных пленок исследовали образцы с меньшими концентрациями (табл. 1). Из полученных данных РЭМ (рис. 3) видно, что форма микрочастиц существенно не меняется, при этом количество наноразмерных частиц с уменьшением концентрации исходного раствора прекурсора падает. Соотношения Ti : V близки в пределах погрешности и равны 5.0 : 1, что совпадает с результатами для эксперимента 1.

При анализе рентгенограмм серии пленок по концентрации исходного прекурсора ванадия выявлена закономерность: с увеличением концентрации раствора более ярко выраженными становятся пики фазы анатаза TiO2 [21-1272]. Нужно отметить, что сигналы, относящиеся к фазе рутила на рентгенограмме образца с самой высокой концентрацией исходного прекурсора, исчезают на рентгенограммах остальных образцов, что может быть связано с низкой интенсивностью рефлексов данной фазы.
Температурная обработка
Порошок и пленка образца с наибольшей концентрацией исходного прекурсора были подвергнуты отжигу в среде аргона при температуре 400°С (4 ч). Условия отжига были выбраны в соответствии с работой [22], где авторы показывают необходимость отжига в вакууме для повышения степени кристалличности фазы и минимизирования образования аморфного покрытия TiO2. Дополнительный отжиг также способствует снятию напряжения в приповерхностном слое пленок, что улучшает их адгезивные и механические свойства.
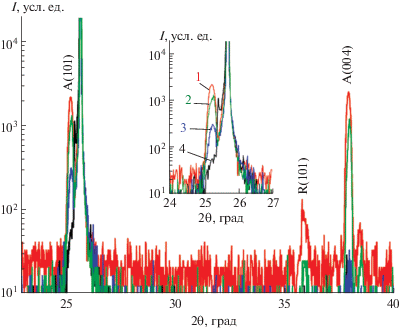
Рис. 4.
Рентгенограммы пленочных образцов, полученных в экспериментах 1–4 с различной концентрацией исходного раствора.
На рис. 5 приведены рентгенограммы порошка и пленки (эксперимент 1) после отжига. Для порошка сдвиг всех линий также сохраняется, однако становится менее заметным. Индицирование рентгенограммы порошка после отжига привело к следующим результатам: а = 3.79, с = 9.46 Å, т.е. параметр с несколько уменьшается по сравнению с фазой анатаза.
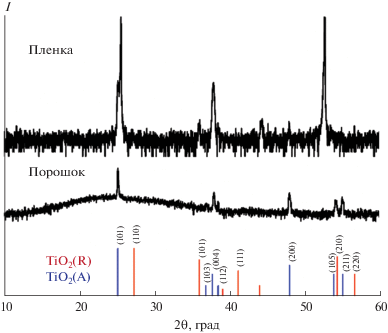
На рентгенограмме пленки после отжига основные сигналы сохраняют свое положение, при этом незначительно изменяется интенсивность рефлексов, в том числе и относящихся к фазе рутила.
Исчезновение микронапряжений продемонстрировано исчезновением сдвига основных рефлексов. В данном случае в качестве стандарта служат рефлексы монокристаллической подложки сапфира.
ЗАКЛЮЧЕНИЕ
В результате гидротермального синтеза при 195оС в течение 6 ч из смеси NH4TiF6 и (NH4)2[(VO)2(C2O4)3(H2O)2] (2 : 1) образуются поликристаллические порошки примерного состава Ti0.83V0.17O2 со структурой анатаза. В аналогичных условиях на поверхности монокристаллического сапфира образуются пленки, текстурированные в направлении (004) и (101). Сдвиг на рентгенограмме поверхности и однородное распределение элементов на поверхности подложек в совокупности свидетельствуют об образовании твердых растворов ванадий-титана и стабилизации ванадия в структуре анатаза. Замечен эффект эпитаксиальной стабилизации, приводящий к образованию рутильной фазы диоксида титана на поверхности r-Al2O3 уже при температуре 195°С.
Список литературы
Sharikov F.Yu., Ivanov V.K., Tret’yakov Yu.D. et al. // Russ. J. Inorg. Chem. 2006. V. 51. Р. 1841. [Шариков Ф.Ю., Иванов В.К., Третьяков Ю.Д. и др. // Журн. неорган. химии. 2006. Т. 51. № 12. С. 1841.] https://doi.org/10.1134/S0036023606120011
Srinivasa R.P., Marinela M., Artemenko A. et al. // Inorg. Chem. 2013. V. 52. P. 4780. https://doi.org/10.1021/ic301201k
Chen S., Dai L., Liu J. et al. // Phys. Chem. Chem. Phys. 2013. V. 15. P. 17537. https://doi.org/10.1039/c3cp52009a
Volkov V.L., Zaharova G.S. et al. // Russ. J. Inorg. Chem. 2006. V. 51. № 6. P. 917. [Волков В.Л., Захарова Г.С., Волкова Е.Г. и др. // Журн. неорган. химии. 2006. Т. 51. № 6. С. 917.] https://doi.org/10.1134/S0036023606060015
Chen Z., Wang X., Qi Y. et al. // ACS Nano. 2016. V. 10. P. 10 237. https://doi.org/10.1021/acsnano.6b05736
Radecka M., Brudnik A., Kulinowski K. et al. // J. Electron. Mater. 2019. V. 48. P. 5481. https://doi.org/10.1007/s11664-019-07266-8
Sun G., Cao X., Yue Y. et al. // Sci. Rep. 2018. V. 8. P. 5342. https://doi.org/10.1038/s41598-018-23412-4
Matsuura Y., Yoshii F., Otsuka T. et al. // J. Eur. Ceram. Soc. 2018. V. 38. P. 5043. https://doi.org/10.1016/j.jeurceramsoc.2018.07.035
Gavrilov A.I., Balakhonov S.V., Churagulov B.R. // Inorg. Mater. 2016. V. 52. P. 1240. https://doi.org/10.1134/S0020168516120049
Wilkinson M., Kafizas A., Bawaked S.M. et al. // ACS Comb. Sci. 2013. V. 15. P. 309. https://doi.org/10.1021/co400027p
Cimieri I., Poelman H., Ryckaert J. et al. // J. Photochem. Photobiol. A: Chem. 2013. V. 263. P. 1. https://doi.org/10.1016/j.jphotochem.2013.04.025
Wu Y., Fan L., Liu Q. et al. // Sci Rep. 2015. V. 5. P. 9328. https://doi.org/10.1038/srep09328
Dhandayuthapani T., Sivakumar R., Ilangovan R. et al. // J. Solid State Electrochem. 2018. V. 22. P. 1825. https://doi.org/10.1007/s10008-018-3888-0
Huang K., Meng Y., Xu X. et al. // J. Phys.: Condens. Matter. 2017. V. 29. P. 355402. https://doi.org/10.1088/1361-648X/aa7707
Wisam J.A., Saja Q.A., Nisreen Z.J. // Silicon. 2018. V. 10. P. 2101. https://doi.org/10.1007/s12633-017-9730-y
Kang Y.B. // J. Eur. Ceram. Soc. 2012. V. 32. P. 3187. https://doi.org/10.1016/j.jeurceramsoc.2012.04.045
Boytsova O.V., Baranchikov A.E., Yapryntsev A.D. et al. // Russ. J. Inorg. Chem. 2018. V. 63. P. 567. https://doi.org/10.1134/S0036023618050029
Lotnyk A., Senz S., Hesse D. // Thin Solid Films. 2007. V. 515. P. 3439. https://doi.org/10.1016/j.tsf.2006.10.106
Makarevich A.M., Sadykov I.I., Sharovarov D.I. et al. // J. Mater. Chem. C. 2015. V. 3. P. 9197. https://doi.org/10.1039/c5tc01811k
Wenda E. // J. Therm. Anal. 1981. V. 20. P. 153. https://doi.org/10.1007/BF01913007
Diebold U. // Surf. Sci. Rep. 2003. V. 48. P. 53. https://doi.org/10.1016/S0167-5729(02)00100-0
Li Y., Ji S., Gao Y. et al. // Sci. Rep. 2013. V. 3. P. 1370. https://doi.org/10.1038/srep01370
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии