

Журнал неорганической химии, 2019, T. 64, № 4, стр. 350-356
Синтез и свойства композитов SnO2@МКЦ, SnO2@C и SnO2/Sn@C
А. Н. Прусов 1, *, С. М. Прусова 1, А. Г. Захаров 1, В. К. Иванов 2, А. В. Базанов 1
1 Институт химии растворов им. Г.А. Крестова РАН
153045 Иваново, ул. Академическая, 1, Россия
2 Институт общей и неорганической химии им. Н.С. Курнакова РАН
119991 Москва, Ленинский пр-т, 31, Россия
* E-mail: anp@isc-ras.ru
Поступила в редакцию 01.10.2018
После доработки 09.10.2018
Принята к публикации 01.11.2018
Аннотация
Предложен метод синтеза композиционного материала SnO2@МКЦ в процессе гидролиза льняной волокнистой целлюлозы водным 10%-ным раствором азотной кислоты, содержащим SnCl2. Полученный композиционный материал SnO2@МКЦ имеет средний размер кристаллитов диоксида олова 3.8 нм. Пиролизом SnO2@МКЦ в инертной среде азота синтезированы углеродные композиты SnO2@C и SnO2/Sn@C. Методами ИК-спектроскопии, рентгенографии, электронной микроскопии, адсорбции азота, термогравиметрии исследованы физико-химические свойства полученных образцов. Установлено, что в процессе пиролиза SnO2@МКЦ при 500οС образуется углеродный композит SnO2@C. Повышение температуры до 800οС способствует образованию композита SnO2/Sn@C. Карботермическое восстановление диоксида олова протекает через образование метастабильного SnO начиная с 679.8οС, образование стабильного кристаллического олова наблюдается с 784.5οС. Удельная площадь поверхности углеродного композита SnO2/Sn@C составляет 110 м2/г.
ВВЕДЕНИЕ
Металлоуглеродные и металлоорганические композиты представляют интерес как новый вид материалов с уникальными свойствами: специфическим адсорбционным сродством и доступностью функциональных возможностей in-pore, однородными структурированными наноразмерными полостями, равномерным, но настраиваемым размером пор, контролируемыми размерами частиц металлов или их оксидов и морфологией [1–9].
Эти материалы перспективны для широкого практического использования: как электронные и оптикоэлектронные устройства, солнечные батареи, газовые сенсоры, гетерогенный катализ [10–18], адсорбенты [19, 20], окислители красителей [21–26], катализаторы горения пороха [27–30], электроды [31–38].
Для синтеза композиционных материалов в качестве матрицы металлов и их оксидов используют биополимеры, включая растительную биомассу [39, 40], волокнистую [41–43] и микрокристаллическую целлюлозу [44], наноцеллюлозу [45] и другие полисахариды [46, 47], углерод [48–50].
Применение целлюлозы в качестве матриц позволяет использовать эффекты самосборки и самоорганизации для синтеза металл- и оксидсодержащих углеродных материалов. Другое важное преимущество этого направления – возможность моделировать состав композита путем смешивания выбранных химических соединений металлов и соответствующих форм целлюлозы (волокнистой, микро- и нанокристаллической).
Использование в качестве матриц активированного угля обусловлено его развитой структурой внутренних пор, что обеспечивает высокую удельную поверхность композита и равномерное распределение наночастиц тонкой дисперсии металлов и их оксидов.
В соответствии с назначением применяют разнообразные технологии изготовления композитов, так как их свойства в значительной степени зависят не только от природы матрицы и оксида металла, но и от способа получения материала. Наиболее известны гидротермальные или сольвотермические методы [51–53], биосинтез [54], золь-гель метод [55], электрохимические [56], метод термического разложения in situ, [57], пиролиз и др. [58]. Каждый из этих методов имеет свои ограничения и недостатки. Заслуживают внимания методы синтеза композитов, позволяющие осуществлять простой контроль над условиями процесса и направленно формировать состав, размер частиц, структуру композитов [49, 50, 52, 59–62].
Целью данной работы является синтез композита на основе льняной волокнистой целлюлозы и SnCl2 ⋅ 2H2O методом in situ, исследование состава и физико-химических свойств продуктов его трансформации в процессе пиролиза в среде азота.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В качестве исходного материала для получения микрокристаллической целлюлозы (МКЦ) использовали льняную волокнистую целлюлозу. Ее химическую обработку проводили следующим образом: 10 г волокнистой льняной целлюлозы после смешения со 100 мл 10%-ного раствора азотной кислоты, содержащего 11 г SnCl2 ⋅ 2H2O, обрабатывали при температуре 95οС в течение 4 ч. Модифицированную МКЦ после фильтрации и промывки дистиллированной водой сушили при 80οС и исследовали ее физико-химические свойства.
Далее полученный образец помещали в кварцевый тигель и прокаливали в горизонтальном проточном реакторе в среде чистого азота. Перед прокаливанием реакционную камеру продували азотом в течение 30 мин. Нагрев до температуры реакции (скорость нагрева 10 град/мин) проводили в токе азота, подаваемого со скоростью 15 см3/мин. Термическую обработку модифицированной целлюлозы осуществляли при 500 и 800οС.
С помощью сканирующего электронного микроскопа Carl Zeiss NVision40 при ускоряющем напряжении 1 кВ с использованием детектора вторичных электронов (SE2) и электронного микроскопа Vega3 Tescan изучали размеры и форму частиц оксида олова в углеродной матрице, ее морфологию.
ИК-спектроскопическое исследование опытных образцов, запрессованных в таблетках КBr, осуществляли на Фурье-спектрометре Vertex 80v с разрешением 0.2 см–1 в диапазоне длин волн 400–4000 см–1. Масса таблеток, их толщина и содержание анализируемого вещества постоянны.
Термогравиметрические исследования опытных образцов проводили на дифференциальном сканирующем калориметре динамического теплового потока DSC 204 F1 Phoenix с использованием термомикровесов TG 209 F1 Iris с разрешением изменения массы 0.1 мкг и нагревом до 1000°С в инертной среде. Обработка данных проведена с использованием программы Thermokinetics Professional SW/KIN.
Кривые широкоуглового рентгеновского рассеяния регистрировали на многофункциональном рентгеновском дифрактометре D8 Advance с использованием CuKα-излучения (λ = 0.1541874 нм). Анализ рентгенограмм проводили с помощью программы Match с использованием базы данных COD.
Удельную поверхность и размеры пор образцов определяли на приборе NOVAtouch™. Удельную поверхность рассчитывали по методу БЭT, а распределение пор по размерам – BJH.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
На рис. 1 приведены ИК-спектр и рентгенограмма композита на основе МКЦ и SnCl2, полученного по методике, представленной выше.
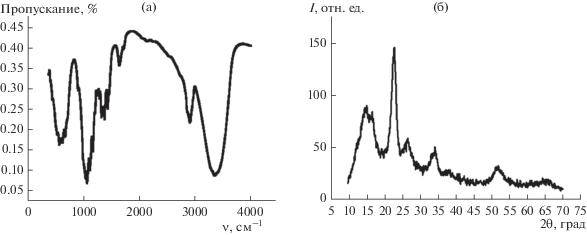
Микрокристаллическая целлюлозная матрица композита сохраняет морфологическую структуру целлюлозы. Об этом свидетельствуют полосы поглощения в области 1100–1300 см–1, принадлежащие целлюлозному основанию, и полоса при 560 см–1, характерная для структурной модификации ЦI (рис. 1а). Полосы поглощения при 609 и 800 см–1 обусловлены образованием SnO2 [17, 63–65].
Интенсивные рефлексы при 2θ = 15.0°, 16.6° и 22.5° на рентгенограмме (рис. 1б) подтверждают сохранение морфологической структуры целлюлозы после химической обработки [66, 67].
Наличие на рентгенограмме рефлексов при 2θ = 26.6°, 33.9°, 51.8° (номер COD 96-900-9083) свидетельствует о присутствии оксида SnO2 (касситерит), имеющего тетрагональную структуру. Полученные результаты согласуются с литературными значениями рефлексов, а именно: 2θ = = 26.6°, 33.8°, 51.8° (номер JCPDS № 41-1445) [64]. Других рефлексов на рентгенограмме не обнаружено, что свидетельствует о чистоте полученного SnO2. Размеры кристаллитов SnO2 рассчитывали по формуле Шеррера [68]:
где λ – длина волны рентгеновского излучения, нм; β – полная ширина в половине максимума рефлекса, рад; θ – угловое положение рефлекса, град.Средний размер кристаллитов SnO2 составляет 3.8 нм. Средний размер кристаллитов диоксида олова, полученного золь-гель методом, равен 3.1 нм [18]. Следовательно, имеет место корреляция полученного результата с литературными данными.
Таким образом, синтез композита SnO2@МКЦ методом in situ представляет собой химический процесс обработки волокнистой льняной целлюлозы, включающий ее гидролиз с получением МКЦ, сорбцию ионов олова целлюлозной матрицей, окисление Sn2+ до Sn4+, образование SnО2.
Расширить функциональные возможности композитов позволяет сочетание разных способов модификации матрицы и множество методов синтеза [69, 70]. В этом плане использование целлюлозной матрицы как потенциального прекурсора углеродного композита открывает широкие перспективы. Целлюлозе свойственна гомогенная пропитка водными растворами солей основного металла. Кроме того, целлюлоза является носителем для однородно диспергированных солей основного металла, их оксидов и наночастиц металлов, полученных из них. Углеродная матрица, получаемая пиролизом льняной МКЦ (800°С) в инертной среде, повторяет ее морфологическую иерархию, что отчетливо иллюстрирует рис. 2. Однако при этом удельная площадь поверхности микрокристаллической целлюлозы составляет 0.3 м2/г, а углеродного образца – 17.4 м2/г.

Композит SnO2@МКЦ подвергали термической обработке в инертной среде в интервале 25–500, 25–800°С и последующей термостабилизации в течение 2 ч. В результате получены композиты SnO2@C и SnO2/Sn@C (рис. 3, 4).
Рис. 3.
Композит SnO2@C, полученный в инертной среде при 500°С: ИК-спектр (а) и электронная микрофотография (б).
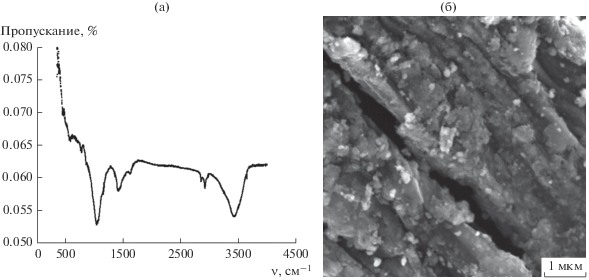
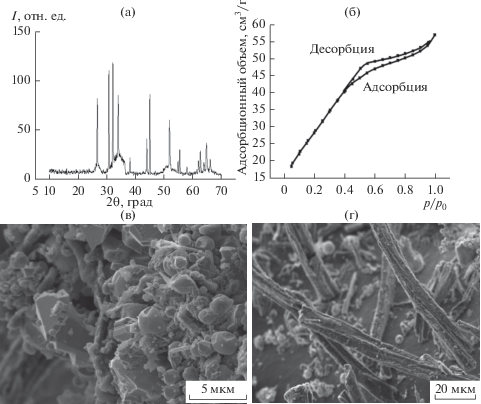
Рис. 4.
Композит SnO2/Sn@C, полученный в инертной среде при 800°С: рентгенограмма (а), изотерма адсорбции-десорбции азота (б) и электронные микрофотографии различного масштаба (в, г).
Как следует из рис. 3а, обработка образца SnO2@МКЦ при 500°С вызывает деструкцию эфирных связей С–О–С, сопровождаемую почти полным исчезновением поглощения в области 1250–400 см–1. Отсутствие полос поглощения, принадлежащих целлюлозной основе, в области 1100–1300 см–1 и полосы при 560 см–1, характерной для целлюлозы структурной модификации I, свидетельствует о переходе целлюлозы в аморфный углерод. Наблюдаемые полосы с максимумами при 609 и 800 см–1 свидетельствуют о наличии SnO2 [17, 63–65]. Полоса поглощения при 1612 см–1 указывает на присутствие O–H-групп молекул воды [64].
Полученный композит SnO2@C представляет собой агломераты частиц SnO2 размером 50–100 нм в углеродной матрице (рис. 3б). Удельная площадь поверхности композита SnO2@C составляет 54.7 м2/г. Таким образом, при термической обработке увеличивается размер агрегатов частиц SnO2. Аналогичные результаты получены в работах [71, 72]. Возможное применение композита SnO2@C в качестве катализатора горения обосновано результатами работ [27–30].
Процесс карботермического восстановления SnO2 до металлического олова в исследованном интервале температур невозможен. Согласно работе [70], для протекания этого процесса необходима температура выше 700οС. Действительно, при 800οС получен углеродный композит, содержащий, наряду с диоксидом олова, кристаллическое олово (рис. 4).
Рентгеноструктурный анализ показал присутствие в углеродной матрице частиц SnO2 (касситерит) и Sn: дифракционные пики при 2θ = 26.6°, 33.9°, 51.8° (номер COD 96-900-9083) и пики при 2θ = 30.7°, 32.1°, 44°, 45° (номер COD 96-900-8571) соответственно (рис. 4а). Средний размер кристаллитов SnO2, рассчитанный по формуле Шеррера [68], составляет 46.9 нм. Полученные результаты свидетельствуют о том, что углеродная матрица при температуре >800°С одновременно выполняет функции носителя и восстанавливающего агента в процессе получения композита SnO2/Sn@C. Кривые интенсивности широкоуглового рентгеновского рассеяния углеродных волокон имеют широкий пик в области углов 2θ = = 28°–38° (CuKα), что свидетельствует о наличии аморфного углерода.
Значительное возрастание интенсивности дифракционных пиков означает увеличение кристаллической фазы частиц. Кроме того, при термической обработке увеличивается размер частиц кристаллитов, что следует из рис. 4в. При этом дисперсный состав неоднороден: наряду с частицами 100–200 нм наблюдаются крупные частицы микронного размера. Поскольку частицы Sn остаются жидкими при температуре реакции из-за низкой температуры плавления (231.9°C), то, вероятно, наличие крупных частиц обусловлено агломерацией металлического олова (рис. 4в).
Полученный композит SnO2/Sn@C имеет типичную изотерму IV типа с петлей гистерезиса, присущей мезопористым материалам, о чем свидетельствует наличие ступеньки на изотерме в области давлений выше диапазона БЭТ (рис. 4б). График БЭТ линеен в диапазоне относительных давлений от 0.05 до 0.35 р/р0, что говорит о хорошей применимости модели БЭТ для расчета удельной площади поверхности полученного композита. Рассчитанная удельная площадь поверхности композита составляет 110 м2/г, общий Vпор = 0.088 см3/г для пор с dпор < 140.4 нм, средний dпор = 3.16 нм. Возрастание удельной поверхности экспериментального образца с ростом температуры обработки можно объяснить воздействием металлических частиц на углеродную матрицу.
Проследить за процессом пиролиза целлюлозы и за трансформацией SnO2 в Sn позволяют полученные термогравиметрические данные.
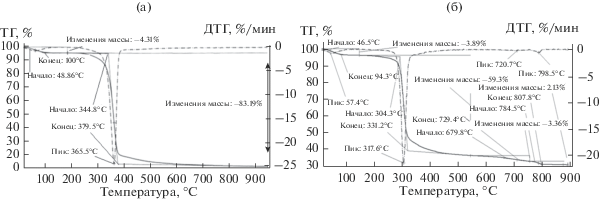
На рис. 5 представлены кривые ТГ и ДТГ для льняной МКЦ (а) и композита SnO2@МКЦ (б). Для обоих образцов первичным является процесс удаления физически связанной воды, протекающий в области до 100οС. Интенсивная стадия термической деструкции льняной МКЦ и SnO2@МКЦ начинается при 344.8 и 304.3°С, а заканчивается при 379.5 и 331.2°С соответственно. Таким образом, при наличии SnO2 в составе композита температура начала интенсивной деградации целлюлозы снижается и сокращается температурный интервал интенсивного разложения матрицы. При этом на кривых ТГ и ДТГ образца с оксидом олова(IV) появляются два пика – при 720.7 и 798.5οС (рис. 5б). Согласно [71–73], восстановление SnO2 углеродом до Sn начинается при 800οС. При этом процесс восстановления оксида олова(IV) до Sn, как предполагают авторы, протекает через стадию восстановления SnO2 в SnO. Как известно, SnO является метастабильным и переходит в стабильное состояние (Sn) при восстановлении углеродом. Таким образом, температура 679.8οС относится к началу стадии восстановления SnO2 в SnO, а 784.5οС – к началу стадии образования Sn.
ЗАКЛЮЧЕНИЕ
Методом in situ проведен синтез композита SnO2@МКЦ, включающий процесс гидролиза волокнистой целлюлозы, сорбцию ионов олова целлюлозной матрицей, окисление Sn2+ до Sn4+, образование SnО2. Установлено, что средний размер кристаллитов SnO2, диспергированных в целлюлозной матрице, составляет 3.8 нм. Пиролизом SnO2@МКЦ в инертной среде азота синтезированы углеродные композиты SnO2@C и SnO2/Sn@C. На основании результатов термогравиметрического исследования высказано предположение, что процесс восстановления SnO2 в Sn протекает через образование метастабильного SnO начиная с 679.8οС. Показано, что в процессе получения углеродного композита SnO2/Sn@C при температуре выше 784.5°С углеродная составляющая композита одновременно выполняет функции носителя и восстанавливающего агента. Полученный композит имеет типичную изотерму адсорбции-десорбции IV типа с петлей гистерезиса, присущей мезопористым материалам. Полученные композиты могут быть использованы в качестве катализаторов.
Список литературы
Tahmasebi E., Masoomi M.Y., Yamini Y., Morsali A. // Inorg. Chem. 2015. V. 54. № 2. P. 425. doi 10.1021/ ic5015384
Eddaoudi M., Kim J., Rosi N. et al. // Science. 2002. V. 295. Issue 5554. P. 469. doi 10.1126/science.1067208
Liu B., Shioyama H., Akita T. et al. // J. Am. Chem. Soc. 2008. V. 130. P. 5390. 5391. doi 10.1021/ja7106146
Wang H., Yu J., Zhao Y. et al. // J. Power Sources. 2013. V. 224. P. 125. doi 10.1016/j.jpowsour.2012.09.051
Sanchez C., Rozes L., Ribot F. et al. // C. R. Chim. 2010. V. 13. P. 3. doi 10.1016/j.crci.2009.06.001
Goncalves G., Marques P.A.A.P., Pinto R.J.B. et al. // Compos. Sci. Technol. 2009. V. 69. № 7–8. P. 1051. doi 10.1016/j.compscitech.2009.01.020
Zhang D., Qi L. // Chem. Commun. 2005. P. 2735. doi 10.1039/b501933h
Menchaca-Nala S., Londoño-Calderóna C.L., Cerruttib P. et al. // Carbohydrate Polymers. 2016. V. 137. P. 726. doi 10.1016/j.carbpol.2015.10.068
Wen Z., Wang Q., Zhang Q., Li J. // Adv. Funct. Mater. 2007. V. 17. P. 2772. doi 10.1002/adfm.200600739
Ansari S.A., Khan M.M., Ansari M.O. et al. // New J. Chem. 2014. V. 38. P. 2462. doi 10.1039/c3nj01488f
Khan M.M., Ansari S.A., Khan M.E. et al. // New J. Chem. 2015. V. 39. P. 2758. doi 10.1039/c4nj02245a
Nouri A., Fakhri A. // Spectrochim. Acta, A: Mol. Biomol. Spectrosc. 2015. V. 138. P. 563. doi 10.1016/j.saa.2014.11.075
Jia X., Liu Y., Wu X. et al. // Appl. Surf. Sci. 2014. V. 311. P. 609. doi 10.1016/j.apsusc.2014.05.118
Ansari S.A., Khan M.M., Ansari M.O. et al. // RSC Adv. 2014. V. 4. P. 26013. doi 10.1039/c4ra03448a
Gupta V.K., Saravanan R., Agarwal S. et al. // J. Mol. Liq. 2017. V. 232. P. 423. doi 10.1016/j.molliq.2017.02.095
Viet P.V., Thi C.M., Hieu L.V. // J. Nanomaterials. 2016. P. 8. doi 10.1155/2016/4231046
Varala R., Narayana V., Kulakarni S.R. et al. // Arabian J. Chem. 2016. V. 9. P. 550. doi 10.1016/j.arabjc.2016.02.015
Oh H.-S., Nong H.N., Strasser P. // Adv. Funct. Mater. 2015. V. 25. P. 1074. doi 10.1002/adfm.201401919
Nilchi A., Dehaghan T.S., Garmarodi S.R. // Desalination. 2013. V. 321. P. 67. doi 10.1016/j.desa1.2012.06.022
Khalameida S., Samsonenko M., Skubiszewska-Zięba J., Zakutevskyy O. // Adsorpt. Sci. Technol. 2017. V. 35. № 9–10. P. 853. doi 10.1177/0263617417722251
Morales J., Petkova G., Cruz M., Caballero A. // J. Power Sources. 2006. V. 158. P. 831. doi 10.1016/j.jpowsour.2005.11.021
Saravanan R., Sacari E., Gracia F. et al. // J. Mol. Liq. 2016. V. 221. P. 1029. doi 10.1016/j.molliq.2016.06.074
Liu S.Z., Ke J., Sun H.Q. et al. // Appl. Catal., B. Environ. 2017. V. 204. P. 358. doi 10.1016/j.apcatb.2016.11.048
Ke J., Liu J., Sun H. et al. // Appl. Catal. B. Environ. 2017. V. 200. P. 47. doi 10.1016/j.apcatb.2016.06.071
Li M., Du H., Kuai L. et al. // Angew. Chem. 2017. V. 129. P. 12823. doi 10.1002/anie.201707647
Nejati‑Moghadam L., Gholamrezaei S., Salavati-Niasari M. et al. // J. Mater. Sci: Mater. Electron. 2017. V. 28. P. 9919. doi 10.1007/s10854-017-6748-2
Денисюк А.П., Демидова Л.А. // Физика горения и взрыва. 2004. Т. 40. № 3. С. 69.
Денисюк А.П., Шепелев Ю.Г., Русин Д.Л., Шумский И.В. // Физика горения и взрыва. 2001. Т. 37. № 2. С. 77.
Сизов В.А., Демидова Л.А., Денисюк А.П. // Успехи в химии и химической технологии. 2015. Т. 29. № 8. С. 16.
Abdel-Ghani N., Elbeih A., Helal F. // Central European J. Energetic Mater. 2016. V. 13. № 2. P. 469. doi 10.22211/cejem/64997
Ma Y., Jia Y., Wang L. et al. // J. Power Sources. 2017. V. 342. P. 921. doi 10.1016/j.jpowsour.2017.01.020
Tan Y.J., Jia Z., J. Sun Z. et al. // J. Mater. Chem. A. 2017. V. 5. P. 24139. doi 10.1039/C7TA08236C
Kong F., Tao S., Qiana B., Gao L. // Ceram. Int. 2018. https://doi.org/10.1016/j.ceramint.2018.08.004.
Sun H.T., Mei L., Liang J. et al. // Science. 2017. V. 356. P. 599. doi 10.1126/science.aam5852
Liu X., Wanga H., Zhang S. et al. // Electrochim. Acta. 2018. V. 292. P. 759. doi 10.1016/j.electacta.2018.09.133
Lin J., Yuana Y., Su Q. et al. // Electrochim. Acta. 2018. V. 292. P. 63. doi 10.1016/j.electacta.2018.09.138
Zhang W., Mao S., Xu J. et al. // Electrochim. Acta. 2018. V. 291. P. 206. doi 10.1016/j.electacta.2018.08.122
Wu K., Shi B., Yin L.Q. et al. // Electrochim. Acta. 2018. V. 291. P. 24. doi 10.1016/j.electacta.2018.09.086
Sevilla M., Sanchís C., Valdés-Solís T. et al. // J. Phys. Chem. C. 2007. V. 111. P. 9749. doi 10.1021/jp072246x-9756
Thompson E., Danks A.E., Bourgeois L. et al. // Green Chem. 2015. V. 117. P. 551. doi 10.1039/C4GC01673D
Sevilla M., Fuertes A.B. // Chem. Phys. Lett. 2010. V. 490. P. 63. doi 10.1016/j.cplett.2010.03.011
Glatzel S., Schnepp Z., Giordano C. // Angew. Chem. Int. Ed. 2013. V. 52. P. 2355. doi 10.1002/ anie.201207693
Hoekstra J., Versluijs-Helder M., Vlietstra E.J. et al. // Chem. Sus. Chem. 2015. V. 8. P. 985. doi 10.1002/cssc.201403364
Hoekstra J., Beale A.M., Soulimani F. et al. // J. Phys. Chem. C. 2015. V. 119. № 19. P. 10653. doi 10.1021/ acs.jpcc.5b00477
Zhu H., Shen F., Luo W. et al. // Nano Energy. 2017. V. 33. P. 37. doi 10.1016/j.nanoen.2017.01.021
Wang H., Yu J., Zhao Y., Guo Q. // J. Power Sources. 2013. V. 224. P. 125. doi 10.1016/j.jpowsour.2012.09.051
Ma B., Huang Y., Zhu C. et al. // J. Alloys Compd. 2016. V. 687. P. 741. doi 10.1016/j.jallcom.2016.06.187
Walsh F.C., Ponce de León C. // Surf. Coat. Technol. 2014. V. 259. P. 676. doi 10.1016/j.surfcoat.2014.10.010
Foresti M.L., Vázquez A., Boury B. // Carbohydrate Polymers. 2017. V. 157. P. 447. doi 10.1016/j.carbpol.2016.09.008
Chesnokov V.V., Chichkan A.S., Luchikhina V.S., Parmon V.N. // Russ. J. Inorg. Chem. 2016. V. 62. № 3. P. 243. [Чесноков В.В., Чичкань А.С., Лучихина В.С., Пармон В.Н. // Журн. неорган. химии. 2016. Т. 61. № 3. С. 288. doi 10.7868/S0044457X16030077]
Oh H.-S., Nong H.N., Strasser P. // Adv. Funct. Mater. 2015. V. 25. P. 1074. doi 10.1002/adfm.201401919
Ghasemi S., Mousavi M., Shamsipur M., Karami H. // Ultrason. Sonochem. 2008. V. 15. № 4. P. 448. https://doi.org/10.1016/j.ultsonch.
Recioa F.J., Herrastia P., Sirés I. et al. // Electrochim. Acta. 2011. V. 56. P. 5158. doi 10.1016/j.electacta.2011.03.054
Khalil A.T., Ovais M., Ullah I. et al. // Arabian J. Chem. 2017. P. 1. doi 10.1016/j.arabjc.2017.08.009
Karami H., Ghamooshi-Ramandi M. // Int. J. Electrochem. Sci. 2013. V. 8. P. 7553.
Karami H., Alipour M. // Int. J. Electrochem. Sci. 2009. V. 4. P. 1511.
Nafees M., Ikram M., Ali1 S. // Appl. Nanosci. 2017. V. 7. P. 399. doi 10.1007/s13204-017-0578-7
Prasad K.H., Vinoth S., Jena P. et al. // Mater. Chem. Phys. 2017. V. 194. P. 188. doi 10.1016/ j.matchemphys.2017.03.040
He J., Kunitake T., Nakao A. // Chem. Mater. 2003. V. 15. № 23. P. 4401. doi 10.1021/cm034720r
Sanchez C., Rozes L., Ribot F. et al. // C. R. Chimie. 2010. V. 13. P. 3. doi 10.1016/j.crci.2009.06.001
Hoekstra J., Beale M., Soulimani F. et al. // Carbon. 2016. V. 107. P. 248. doi 10.1016/j.carbon.2016.05.065
Zakharov A.G., Prusov A.N., Prusova S.М. et al. // Fibre Chemistry. 2015. № 4. [Захаров А.Г., Прусов А.Н., Прусова С.М. и др. // Химические волокна. 2015. № 4. С. 63.]
Vaughan T., Seo C.W., Marshall W.E. // Bioresour. Technol. 2001. V. 78. P. 133. doi 10.1016/S0960-8524(01)00007-4
Viet P.V., Thi C.M., Hieu L.V. // J. Nanomater. 2016. P. 1. doi 10.1155/2016/4231046
Majumder S. // Materials Science-Poland. 2009. V. 27. P. 123.
Zhang W., Yi Z.L., Huang J.F. et al. // Bioresour. Technol. 2013. V. 130. P. 30. doi 10.1016/j.biortech.2012.12.029
Wada M., Okano T. // Cellulose. 2001. V. 8. P. 183. doi 10.1023/A:1013196220602
Patil G.E., Kajale D.D., Gaikwad V.B., Jain G.H. // Int. Nano Lett. 2012. V. 2. P. 46. doi 10.5402/2012/275872
Huang J., Matsunaga N., Shimanoe K. et al. // Chem. Mater. 2005. V. 17. № 13. P. 3513. doi 10.1021/ cm047819m
Ma H., Teng K., Fu Y. et al. // Energy Environ. Sci. 2011. V. 4. P. 3067.
Harangi Z., Kékesi T. // Mater. Sci. Eng. 2014. V. 39. P. 13.
Padilla R., Sohn H.Y. // Metallurg. Trans. B. 1979. V. 10. P. 109. doi 10.1007/BF02653980
Mitchell A.R., Parker R.H. // Miner. Eng. 1988. V. 1. P. 53.
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии