

Журнал неорганической химии, 2019, T. 64, № 3, стр. 288-295
Теоретическое исследование изомеров допированных кластеров L2Al42 с допантами L переходных элементов внутри и на поверхности алюминиевого каркаса
О. П. Чаркин 1, *, Н. М. Клименко 2
1 Институт проблем химической физики РАН
Черноголовка, Московской обл.,
пр-т Академика Семенова, 1, Россия
2 Московский государственный технологический университет им. М.В. Ломоносова
119571 Москва, пр-т Вернадского, 86, Россия
* E-mail: charkin@icp ac.ru
Поступила в редакцию 25.06.2018
После доработки 15.08.2018
Принята к публикации 05.07.2018
Аннотация
В рамках метода функционала плотности выполнены расчеты структуры и стабильности внутренних и внешних изомеров у семейства наноразмерных кластеров L2Al42 с атомами допантов переходных элементов L внутри и на поверхности алюминиевого каркаса. Установлено, что для всех кластеров с L = Sc−Cu предпочтительны изомеры с допантами во внутренних (закрытых) позициях в ядре кластера. Переход допантов на поверхностные позиции и превращение их в открытые каталитические центры требуют затрат энергии ∼15−25 ккал/моль. Впервые предсказана высокая стабильность и возможность существования высокоспиновых кластеров типа L5Al39 с пятью допантами, расположенными в предпочтительных аксиальных и экваториальных внутренних позициях. Результаты могут быть полезными для теоретического прогнозирования и моделирования более крупных молекулярных алюминиевых сплавов c более высоким содержанием допантов L.
В последние годы была опубликована серия работ, посвященных квантово-химическим расчетам электронной и геометрической структуры, стабильности, спектроскопических и других молекулярных характеристик алюминиевых кластеров, допированных атомами других элементов [1–8]. Авторы изучали зависимость структуры и свойств нейтральных и заряженных систем от размеров и формы алюминиевых кластеров [1], исследовали каталитические свойства микросплавов переходных 3d-металлов в поверхностной области алюминиевого каркаса [2], моделировали механизмы реакций взаимодействия анионов борзамещенных алюминидов с молекулярным кислородом [3], активации молекулярного азота с использованием Si- и P-допированных кластеров алюминия [4] и других процессов. В ряде работ рассчитаны поверхности потенциальной энергии и исследованы механизмы диссоциативного присоединения молекулярного водорода к алюминиевым кластерам, допированным атомом магния, 3d- и 4d-переходных металлов. Аналогичные расчеты были выполнены для реакций присоединения молекулы Н2 к Mg-допированному кластеру MgAl12 [6]. Структурные особенности алюминиевых сплавов с переходными металлами изучены в [7]. В [8] бинарные кластеры LAl12 исследованы методами масс-спектрометрии и фотоэлектронной спектроскопии с интерпретацией результатов в рамках концепции “суператомов”.
Как правило, подобные расчеты выполняли с помощью методов теории функционала плотности (ТФП/DFT) и они посвящены небольшим кластерам типа LAl12 с одним допантом L и икосаэдрической структурой каркаса, в которой все атомы занимают поверхностные позиции, за исключением одного – в центре икосаэдра. Недавно (см. работу [9] и библиографию в ней) DFT-подход был распространен на более сложные алюминиевые кластеры С2Al42, C6Al38, C12Al32, Si6Al38, N6Al38 и P6Al38 с пятиатомным внутренним ядром и несколькими допантами. В этих работах прослеживаются тенденции изменения структуры и относительной стабильности внутренних и внешних изомеров (c допантами в составе внутреннего ядра и на поверхности кластера соответственно) при изменении допантов L в ряду углерод–кремний–азот–фосфор. Этот вопрос связан с общей проблемой влияния примесей и легирующих добавок на структуру и свойства наноразмерных частиц. Важно также понять, в каких случаях предпочтительным сохраняется рассеянное распределение допантов по всему объему кластера в виде разделенных атомов (ионов) и в каких при увеличении числа допантов может появиться возможность микрокластеризации допантов с образованием их ковалентно-связанных двухатомных или более сложных частиц типа [Lm]. Этот вопрос связан с проблемой поиска пограничных областей в стехиометрическом соотношении допанта и основного металла, ниже которых атомы (ионы) допантов рассеиваются стохастически и выше которых становится существенной кластеризация допанта вплоть до сегрегации компонентов.
В нашей предыдущей работе [10] выполнены DFT-расчеты изомеров допированных кластеров L2Al42, полученных из алюминиевого кластера Al44 при замещении двух его атомов Al атомами легких непереходных sp-элементов, Cu, Zn и H в различных позициях на поверхности и внутри алюминиевого каркаса. Нас интересовало, как меняется структура и относительная энергетическая стабильность внутренних и внешних изомеров при изменении допанта L вдоль второго и третьего непереходных периодов и при переходе от 2s2p-элементов к их 3s3p-аналогам. Расчеты [10] свидетельствовали, что для большинства допантов sp-элементов и атома Н предпочтительны позиции допантов либо на поверхности, либо в подповерхностном (промежуточном) пространстве, в котором допант L связан с атомами алюминия, расположенными как на поверхности, так и во внутреннем ядре каркаса. С другой стороны, для легких элементов второго периода (L = Ве и В) и элементов конца 3d-периода (L = Сu и Zn) самыми выгодными и более предпочтительными являются внутренние позиции допантов. Результаты сравнивали с данными предыдущих DFT-расчетов изомеров кластеров LAl12 с теми же допантами L [11]. Рассмотрено влияние размера алюминиевого каркаса на относительную энергетическую стабильность поверхностных и внутренних позиций допантов.
В настоящем сообщении, являющемся продолжением [10], DFT-расчеты распространены на родственные кластеры L2Al42 с допантами элементов первого и второго переходных периодов (L = Sc−Ni, Zr, Mo и Pd), расположенными в позициях внутри и на поверхности каркаса. Основное внимание фокусируется на энергетических характеристиках внутренних и внешних изомеров и тенденциях их поведения при изменении допантов вдоль переходных периодов. Вопрос об относительной стабильности внутренних и внешних изомеров важен при рассмотрении каталитической активности допантов d- и f-переходных металлов, где максимальная активность соответствует открытым поверхностным позициям, в то время как во внутренних позициях допант пространственно закрыт атомами алюминия и не активен. Представляло также интерес оценить стабильность изомеров к распаду с отщеплением двух атомов допанта
(1)
${{{\text{L}}}_{{\text{2}}}}{\text{A}}{{{\text{l}}}_{{{\text{42}}}}} \to {\text{A}}{{{\text{l}}}_{{42}}} + 2{\text{L}} + {{Е }_{{{\text{д и с }}}}}$Экспериментальные исследования подобных проблем на уровне наноразмерных частиц сталкиваются с трудностями, и значительную помощь здесь могут оказать модельные квантово-химические подходы и расчеты, которые в некоторых случаях оказываются единственным источником полезной информации. Из-за приближенного характера метода DFT основное внимание уделяется не столько абсолютным значениям рассчитанных величин, сколько их относительным изменениям и тенденциям в родственных рядах кластеров L2Al42. Рассмотрение имеет модельный и предсказательный характер.
Работа состоит из нескольких частей. В первой части на примере тестовых кластеров Ti2Al42, Ni2Al42, Zn2Al42, Zr2Al42 и Pd2Al42 оптимизированы те же восемь базовых стартовых структур, которые подробно исследовались в [10] и изображены на рис. 1. Две из них – (${\mathbf{a}}_{{\mathbf{I}}}^{ + },{\mathbf{a}}_{{\mathbf{I}}}^{ - }$) и (${{{\mathbf{e}}}_{0}},{\mathbf{e}}_{0}^{{\text{'}}}$) – соответствуют внутренним, а остальные шесть – (${\mathbf{v}}_{{{\mathbf{II}}}}^{ + },{\mathbf{v}}_{{{\mathbf{II}}}}^{ - }$), (${\mathbf{v}}_{{\mathbf{I}}}^{ + },{\mathbf{v}}_{{\mathbf{I}}}^{ - }$), (${{{\mathbf{v}}}_{0}},{\mathbf{v}}_{0}^{{\text{'}}}$), (${\mathbf{m}}_{{{\mathbf{II}}}}^{ + },{\mathbf{m}}_{{{\mathbf{II}}}}^{ - }$), (${\mathbf{m}}_{{\mathbf{I}}}^{ + },{\mathbf{m}}_{{\mathbf{I}}}^{ - }$) и (${{{\mathbf{m}}}_{0}},{\mathbf{m}}_{0}^{{\text{'}}}$) − поверхностным изомерам. (Здесь используются обозначения позиций v, m, a, e и слоев 0, I± и II±, принятые в [10]). Относительные энергии равновесных структур, оптимизированных в настоящей работе, приведены в табл. 1. Расчеты ставили целью выявить в каждой из этих двух групп энергетически наиболее выгодные изомеры. Показано, что для большинства рассмотренных в первой группе кластеров наиболее выгодными являются близколежащие (аксиальный и экваториальный) внутренние изомеры (${\mathbf{a}}_{{\mathbf{I}}}^{ + },{\mathbf{a}}_{{\mathbf{I}}}^{ - }$) и (${{{\mathbf{e}}}_{0}},{\mathbf{e}}_{0}^{{\text{'}}}$), а во второй группе – изомер (${\mathbf{m}}_{{\mathbf{I}}}^{ + },{\mathbf{m}}_{{\mathbf{I}}}^{ - }$) с допантами в промежуточных слоях (см. ниже).
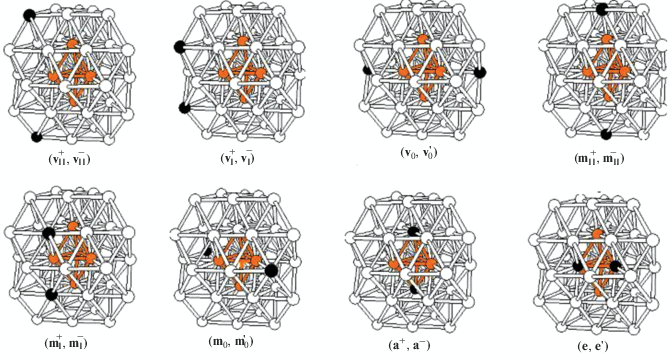
Таблица 1.
Относительные энергии Еотн ключевых поверхностных и внутренних изомеров кластеров L(3d)2Al42, допированных атомами переходных металлов 3d-периода
| Параметр | Относительная энергия, Еотн, ккал/моль* | |||||
|---|---|---|---|---|---|---|
| Ti2Al42 (S = 1) |
Mn2Al42 (S = 11) |
Ni2Al42 (S = 1) |
Zn2Al42 (S = 1) |
Zr2Al42 (S = 1) |
Pd2Al42 (S = 1) |
|
| (${\mathbf{v}}_{{{\mathbf{II}}}}^{ + },{\mathbf{v}}_{{{\mathbf{II}}}}^{ - }$) | 47 | 34 | 35 | 29 | 40 | 9 |
| (${\mathbf{v}}_{{\mathbf{I}}}^{ + },{\mathbf{v}}_{{\mathbf{I}}}^{ - }$) | 30 | 36 | 46 | 24 | 29 | 18 |
| (${{{\mathbf{v}}}_{0}},{\text{ }}{\mathbf{v}}_{0}^{{\text{'}}}$) | 26 | 44 | 42 | 14 | 14 | 0 |
| (${\mathbf{m}}_{{{\mathbf{II}}}}^{ + },{\mathbf{m}}_{{{\mathbf{II}}}}^{ - }$) | 26 | 19 | 55 | 22 | 24 | 16 |
| (${\mathbf{m}}_{{\mathbf{I}}}^{ + },{\mathbf{m}}_{{\mathbf{I}}}^{ - }$) | 15 | 8 | ∼41 | 10 | 0 | |
| (${{{\mathbf{m}}}_{0}},{\text{ }}{\mathbf{m}}_{0}^{{\text{'}}}$) | 23 | 10 | 47 | 11 | 20 | |
| (${\mathbf{a}}_{{\mathbf{I}}}^{ + },{\mathbf{a}}_{{\mathbf{I}}}^{ - }$) | 2 | 0 | 0 | 0 | 9 | 0 |
| (${{{\mathbf{e}}}_{0}},{\text{ }}{\mathbf{e}}_{0}^{{\text{'}}}$) | 0 | 35 | 16 | 4 | 0 | |
* Расчеты Еотн выполнены в приближении B3LYP/6-31G* при геометрии, оптимизированной на уровне B3LYP/6-31G для наиболее выгодных высокоспинового состояния у изомеров Mn2Al42 (S = 11) и синглетных состояний у остальных кластеров. За начало отсчета выбраны энергии наиболее выгодных внутренних изомеров (${\mathbf{a}}_{{\mathbf{I}}}^{ + },{\mathbf{a}}_{{\mathbf{I}}}^{ - }$) и (${{{\mathbf{e}}}_{0}},{\text{ }}{\mathbf{e}}_{0}^{{\text{'}}}$).
Во второй части оба предпочтительных изомера – (${\mathbf{a}}_{{\mathbf{I}}}^{ + },{\mathbf{a}}_{{\mathbf{I}}}^{ - }$) и (${\mathbf{m}}_{{\mathbf{I}}}^{ + },{\mathbf{m}}_{{\mathbf{I}}}^{ - }$) – были оптимизированы для всей серии кластеров L2Al42 с допантами переходных металлов от Sc до Zn. Оптимизированные структуры изомеров (${\mathbf{a}}_{{\mathbf{I}}}^{ + },{\mathbf{a}}_{{\mathbf{I}}}^{ - }$) и (${\mathbf{m}}_{{\mathbf{I}}}^{ + },{\mathbf{m}}_{{\mathbf{I}}}^{ - }$) изображены на рис. 2. В табл. 2 рассмотрены тенденции поведения относительных энергий ΔEотн изомеров при изменении L вдоль 3d-ряда. Как показали расчеты, для всех рассмотренных кластеров закрытые изомеры с допантами во внутренних позициях ${\mathbf{a}}_{{\mathbf{I}}}^{ + }$ и e0 существенно более выгодны по сравнению с открытыми аналогами (см. ниже).
Рис. 2.
Оптимизированные структуры наиболее выгодных внутреннего (${\mathbf{a}}_{{\mathbf{I}}}^{ + },{\mathbf{a}}_{{\mathbf{I}}}^{ - }$) и поверхностного (${\mathbf{m}}_{{\mathbf{I}}}^{ + },{\mathbf{m}}_{{\mathbf{I}}}^{ - }$) изомеров кластеров L2Al42.
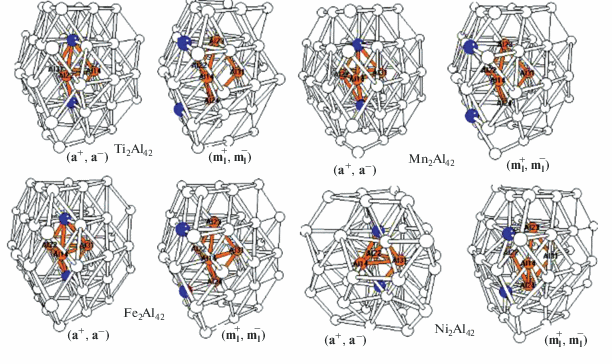
Таблица 2.
Рассчитанные характеристики низших внутреннего и поверхностного изомеров алюминиевых кластеров L2Al42, допированных атомами элементов 3d-периода, ккал/моль
| Параметр* | Al42Ti2 (S = 5) |
Al42V2 (S = 7) |
Al42Cr2 (S = 9) |
Al42Mn2 (S = 11) |
Al42Fe2 (S = 9) |
Al42Co2 (S = 7) |
Al42Ni2 ** | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ${\mathbf{a}}_{{\mathbf{I}}}^{ + },{\mathbf{a}}_{{\mathbf{I}}}^{ - }$ | ${\mathbf{m}}_{{\mathbf{I}}}^{ + },{\mathbf{m}}_{{\mathbf{I}}}^{ - }$ | ${\mathbf{a}}_{{\mathbf{I}}}^{ + },{\mathbf{a}}_{{\mathbf{I}}}^{ - }$ | ${\mathbf{m}}_{{\mathbf{I}}}^{ + },{\mathbf{m}}_{{\mathbf{I}}}^{ - }$ | ${\mathbf{a}}_{{\mathbf{I}}}^{ + },{\mathbf{a}}_{{\mathbf{I}}}^{ - }$ | ${\mathbf{m}}_{{\mathbf{I}}}^{ + },{\mathbf{m}}_{{\mathbf{I}}}^{ - }$ | ${\mathbf{a}}_{{\mathbf{I}}}^{ + },{\mathbf{a}}_{{\mathbf{I}}}^{ - }$ | ${\mathbf{m}}_{{\mathbf{I}}}^{ + },{\mathbf{m}}_{{\mathbf{I}}}^{ - }$ | ${\mathbf{a}}_{{\mathbf{I}}}^{ + },{\mathbf{a}}_{{\mathbf{I}}}^{ - }$ | ${\mathbf{m}}_{{\mathbf{I}}}^{ + },{\mathbf{m}}_{{\mathbf{I}}}^{ - }$ | ${\mathbf{a}}_{{\mathbf{I}}}^{ + },{\mathbf{a}}_{{\mathbf{I}}}^{ - }$ | ${\mathbf{m}}_{{\mathbf{I}}}^{ + },{\mathbf{m}}_{{\mathbf{I}}}^{ - }$ | ${\mathbf{a}}_{{\mathbf{I}}}^{ + },{\mathbf{a}}_{{\mathbf{I}}}^{ - }$ | ${\mathbf{m}}_{{\mathbf{I}}}^{ + },{\mathbf{m}}_{{\mathbf{I}}}^{ - }$ | |
| R(LL) | 4.53 | 4.51 | 4.50 | 4.53 | 4.53 | 4.48 | 4.38 | 4.72 | 4.26 | 4.31 | 4.44 | 4.12 | 4.04 | 3.96 |
| ν(LL) | 353 | 360 | 378 | 385 | 387 | 403 | 390 | |||||||
| ρ(2L) | 2.6 | 3.4 | 5.8 | 6.2 | 7.6 | 9.0 | 9.0 | 9.6 | 6.4 | 7.2 | 2.6 | 4.2 | 0.2 | 2.0 |
| ρ(Al42) | 1.4 | 0.6 | 0.5 | –0.2 | 0.4 | –1.1 | 1.1 | 0.3 | 1.5 | 0.7 | 3.5 | 1.7 | 3.8 | 2.0 |
| Еотн | 0 | 12.5 | 0 | 22.0 | 0 | 14.0 | 0 | 3.8 | 0 | 23.2 | 0 | 20.7 | 0 | 41 |
| Eдис(1) | 64.6 | 62.7 | 80.9 | –10 | 41.4 | 69.7 | ∼100 | |||||||
* Расстояние между допантами R(LL) и частота колебания ν(LL) выражены в Å и см–1 соответственно. ρ(L) − спиновая плотность на атоме допанта, а ρ(Al42) − суммарная спиновая плотность алюминиевого каркаса Al42 (в долях е). Еотн − относительные энергии изомеров ${\mathbf{a}}_{{\mathbf{I}}}^{ + },{\mathbf{a}}_{{\mathbf{I}}}^{ - }$ и ${\mathbf{m}}_{{\mathbf{I}}}^{ + },{\mathbf{m}}_{{\mathbf{I}}}^{ - }$; Eдис(1) – энергия распада допированного кластера по схеме L 2Al42 → Al42 + 2L, приходящаяся на один атом L. Энергии Еотн и Eдис(1) выражены в ккал/моль. ** У кластера Al42Ni2 предпочтительный синглет и соседние триплет и квинтет расположены тесной группой в пределах нескольких ккал/моль.
В третьем разделе рассчитана серия новых кластеров L5Al39 с пятью допантами (L = Sc−Ni). Их оптимизированные структуры, в которых допанты заполняют все пять самых выгодных внутренних позиций (2а + 3е) в форме искаженного бипирамидального ядра (сердечника), встроенного во внутреннюю полость каркаса Al39, изображены на рис. 3. Согласно расчетам, этому изомеру отвечает самая низкая полная энергия Еполн у всех кластеров L5Al39 с допантами переходных металлов. В табл. 3 приведены относительные энергии низколежащих мультиплетов и спиновые плотности на сердечнике ρ(L5) и каркасе ρ(Al39), рассчитанные для систем с разными допантами L.
Рис. 3.
Оптимизированные структуры кластеров L5@Al39 c бипирамидальным магнитным сердечником L5 внутри алюминиевого каркаса Al39.
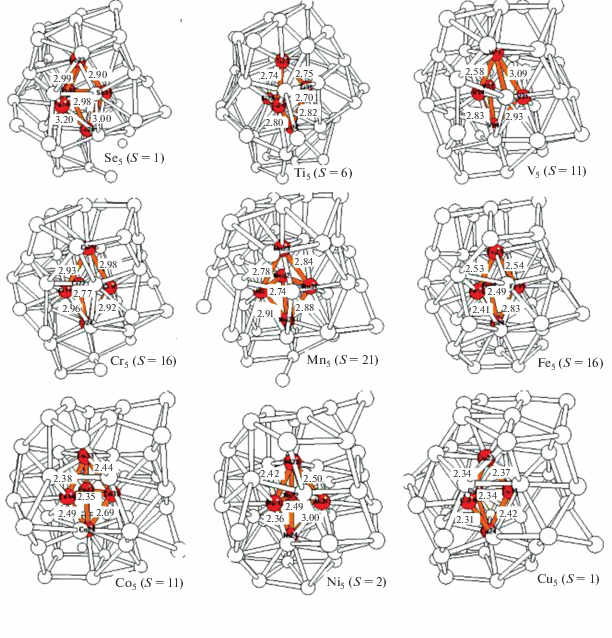
Таблица 3.
Относительные энергии ΔЕотн (в ккал/моль) и распределение спиновой плотности ρ между сердечником L5 и каркасом Al39 (в долях е) у низколежащих мультиплетов кластеров L5@Al39 (1)*
| Кластер L5@Al39 |
Спин, S |
ΔЕотн, ккал/моль |
ρ(L5), е |
ρ(Al39), е |
Кластер L5@Al39 |
Спин, S |
ΔЕотн, ккал/моль |
ρ(L5), е |
ρ(Al39) е |
|---|---|---|---|---|---|---|---|---|---|
| Sc5@Al39 | 1 3 5 |
0 5.3 12.9 |
– 0.7 1.3 |
– 1.3 2.7 |
Fe5@Al39 | 16 12 8 |
0 14.9 38.0 |
14.3 13.8 13.6 |
0.7 –2.8 –6.6 |
| Ti5@Al39 | 6 4 2 |
0 0.6 2.4 |
2.8 2.0 1.5 |
2.2 1.0 –1.5 |
Co5@Al39 | 11 7 3 |
0 17.7 26.4 |
8.5 7.4 2.1 |
1.5 –1.4 –0.1 |
| V5@Al39 | 11 7 3 |
0 2.3 16.9 |
7.4 3.9 2.4 |
2.6 2.1 –0.4 |
Ni5@Al39 | 2 4 6 |
0 8.7 16.1 |
0.7 0.7 1.7 |
0.3 2.3 3.3 |
| Cr5@Al39 | 16 12 8 |
0 18.8 51.8 |
17.7 17.0 16.1 |
–2.7 –6.0 –9.1 |
Cu5@Al39 | 1 3 5 |
0 11.7 30.0 |
– 0.3 0.5 |
– 1.7 3.5 |
| Mn5@Al39 | 21 17 13 |
0 10.9 33.6 |
21.6 20.0 19.3 |
–1.6 –4.0 –7.3 |
Zn5@All39 | 2 4 6 |
0 10.1 27.1 |
0.1 0.4 0.5 |
0.9 2.6 4.5 |
Все расчеты кластеров с допантами 3d-элементов выполнены с помощью программы GAUSSIAN-09 [12] в рамках приближения B3LYP [13, 14] с базисами 6-31G (оптимизация геометрии и расчеты частот колебаний) и 6-31G* (повторная оптимизация геометрии и расчет энергетических характеристик). В расчетах кластеров с допантами 4d-элементов использовали смешанный базис Gen = LANL2DZ(L) + 6-31G(Al).
Отметим, что расчеты кластеров с участием атомов d-металлов с открытыми оболочками могут представлять сложную задачу из-за высокой плотности и пересечений низколежащих термов с различной мультиплетностью, из-за присутствия термов с одинаковой мультиплетностью, но с разным распределением электронной и спиновой плотности, из-за многодетерминантного характера волновых функций и др. Здесь мы ограничились оптимизацией высокоспиновых мультиплетов c S = Smax и расчетом вертикальных состояний с мультплетностями S = Smax − 2, Smax − 4, Smax − 6 и т.д. при геометрии, оптимизированной для S = = Smax. В рамках приближения B3LYP высокоспиновые состояния (S = Smax) с однотерминантными функциями должны описываться точнее, чем низкоспиновые, и в настоящей работе основное внимание концентрировалось на относительных изменениях рассчитанных величин в рядах родственных систем.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Изомерия кластеров Ti2Al42, Mn2Al42, Ni2Al42, Zr2Al42 и Pd2Al42. Из табл. 1 следует, что в рамках приближения B3LYP/6-31G* кластерам Ti2Al42 и Mn2Al42 отвечают наиболее выгодные высокоспиновые состояния с мультиплетностями Smax = 5 и 11. Рядом с ними (в пределах 5−8 и 7−12 ккал/моль) располагаются состояния с более низкими значениями S = 3 и 1, 9 и 7 соответственно. У Ni2Al42 различия в энергиях синглета, триплета и квинтета не превышают ∼5 ккал/моль, причем синглет и триплет практически вырождены. У состояний кластеров Ti2Al42 и Mn2Al42 с Smax спиновые плотности на атомах 3d-металлов варьируют в пределах ∼1.3−1.6 и ∼4.5−4.8 e соответственно. На качественном уровне они ближе всего соответствуют двухвалентному состоянию атомов титана и марганца, где они сохраняют два и пять неспаренных электронов с параллельными спинами. С другой стороны, в триплетном состоянии Ni2Al42 спиновая плотность ρ(Ni) не превышает десятых долей е, так что состояние атомов никеля в этом кластере близко к нульвалентному.
При уменьшении суммарной мультиплетности кластера, например при переходе от Smax = 11 к S = 9, 7 и 5 у Mn2Al42 (табл. 2), спиновая плотность допанта ρ(Mn) уменьшается медленно и не опускается ниже 4.3−4.5 е. Этот результат свидетельствует о том, что уменьшение S связано в основном с перераспределением спиновой плотности каркаса ρ(Al42). При этом спины допантов, с одной стороны, и спины каркаса Al42 – с другой, могут иметь противоположное направление (антиферромагнитный эффект) и частично компенсировать друг друга. Похожая картина имеет место при возбуждении термов у внутреннего изомера Ni2Al42 (${\mathbf{а }}_{{\mathbf{I}}}^{ + },{\mathbf{а }}_{{\mathbf{I}}}^{ - }$). В этом случае спиновая плотность ρ(Ni) остается малой как в триплетном (0.07 е), так и в квинтетном (0.18 е) состоянии, так что увеличение мультиплетности S этого кластера почти целиком связано с увеличением спиновой плотности каркаса ρ(Al42).
Из табл. 1 следует, что для всех тестовых кластеров явно предпочтительными являются внутренние изомеры с допантами в аксиальных (${\mathbf{а }}_{{\mathbf{I}}}^{ + },{\mathbf{а }}_{{\mathbf{I}}}^{ - }$) и экваториальных (e, e') позициях ядра. У Ti2Al42 эти изомеры близки друг к другу в пределах нескольких ккал/моль, а у Mn2Al42 и Ni2Al42 экваториальный изомер значительно менее выгоден, чем аксиальный. Среди поверхностных изомеров предпочтительна структура ${\mathbf{m}}_{{\mathbf{I}}}^{ + },{\mathbf{m}}_{{\mathbf{I}}}^{ - }$, но и она остается на ∼15 (Ti2Al42), ∼8 (Mn2Al42) и ∼41 (Ni2Al42) ккал/моль менее выгодной, чем ${\mathbf{а }}_{{\mathbf{I}}}^{ + },{\mathbf{а }}_{{\mathbf{I}}}^{ - }$. Аналогичный результат получен в расчетах кластеров Cu2Al42 и Zn2Al42, у которых внутренний изомер выгоднее поверхностного на ∼25 и ∼22 ккал/моль [10].
В отличие от систем L2Al42 с допантами 3d-элементов, для аналогов с допантами 4d-элементов (в табл. 1 приведены данные расчетов для кластеров Zr2Al42 и Pd2Al42) предпочтительны низкоспиновые (синглетные) состояния. Возбуждение ближайших триплетных и квинтетных состояний из основного синглетного связано с затратами энергии ∼4−6 и ∼10−12 ккал/моль соответственно.
Сравнение внутреннего (${\mathbf{а }}_{{\mathbf{I}}}^{ + },{\mathbf{а }}_{{\mathbf{I}}}^{ - }$) и поверхностного (${\mathbf{m}}_{{\mathbf{I}}}^{ + },{\mathbf{m}}_{{\mathbf{I}}}^{ - }$) изомеров кластеров L2Al42 (L = Ti−Ni). Рассмотрим тенденции поведения энергетических, структурных и электронных характеристик низших внутреннего и поверхностного изомеров у высокоспиновых кластеров L2Al42 при изменении допанта вдоль 3d-периода.
Анализ относительных энергий (Еотн, табл. 2) показывает, что для всего семейства этих кластеров наиболее выгодны аксиальный и близко расположенный экваториальный внутренние изомеры ${\mathbf{а }}_{{\mathbf{I}}}^{ + },{\mathbf{а }}_{{\mathbf{I}}}^{ - }$ и eO,eO, в которых оба допанта полностью экранированы соседними поверхностными атомами алюминия. Ближайший изомер с допантами в открытых поверхностных позициях ${\mathbf{m}}_{{\mathbf{I}}}^{ + },{\mathbf{m}}_{{\mathbf{I}}}^{ - }$ лежит выше на ∼13−23 ккал/моль, а остальные поверхностные структуры еще менее выгодны. Во всех случаях перемещение (промотирование) допантов из закрытых внутренних на открытые (каталитически активные) поверхностные позиции должно быть связано с существенными затратами энергии.
Энергии Едис распада по схеме
(1)
${{{\text{L}}}_{{\text{2}}}}{\text{A}}{{{\text{l}}}_{{{\text{42}}}}}{\text{(}}{\mathbf{a}}_{{\mathbf{I}}}^{ + }{\text{,}}{\mathbf{a}}_{{\mathbf{I}}}^{ - }{\text{)}} \to {\text{A}}{{{\text{l}}}_{{{\text{42}}}}} + {\text{2L,}}$Из табл. 2 видно, что расстояния R(LL) между допантами у обоих изомеров варьируют в пределах ∼4.0−4.5 Å и имеют тенденцию к уменьшению с увеличением порядкового номера атома М и его атомного радиуса. Из анализа спиновой плотности ρ следует, что допанты V, Cr и Mn в кластерах сохраняют ∼3, ∼4 и ∼5 неспаренных электронов с параллельными спинами, а плотность алюминиевого каркаса ρ(Al42) при этом не превышает 0.5−1.1 е. В конце 3d-периода плотность ρ(Al42) увеличивается до ∼3.8 е, свидетельствуя, в частности, о том, что у внутреннего изомера ${\mathbf{а }}_{{\mathbf{I}}}^{ + },{\mathbf{а }}_{{\mathbf{I}}}^{ - }$ кластера Ni2Al42 в квинтетном состоянии (S = 5) спиновая плотность практически полностью локализуется на алюминиевом каркасе, а атомы никеля остаются близкими к нульвалентным.
Помимо структур, изображенных на рис. 2, нами была рассчитана серия поверхностных изомеров с двумя соседними атомами L, которые располагаются на общих ребрах каркаса и могут играть роль двухцентровых каталитических центров. Их подробное рассмотрение будет дано позднее при изучении механизмов присоединения к ним молекул Н2 и углеводородов к “активным” ребрам Ti−Ti, Ni−Ni и др. Здесь важен вывод, что все изомеры L2Al42 с допантами на общем ребре менее выгодны по сравнению с внутренним изомером ${\mathbf{а }}_{{\mathbf{I}}}^{ + },{\mathbf{а }}_{{\mathbf{I}}}^{ - }$ на 20−35 ккал/моль. Аксиальные и экваториальные позиции а и е внутреннего ядра остаются предпочтительными у всех рассмотренных выше соединений.
Кластеры L5@Al39. В заключение мы рассчитали структуру и свойства еще одного семейства кластеров типа L5@Al39 с пятью атомами переходных металлов, которые занимают самые выгодные позиции (2а + 3е) и образуют бипирамидальный сердечник L5, встроенный во внутреннюю полость каркаса Al39 (рис. 3). В табл. 3 сравниваются относительные энергии ΔEотн низших мультиплетов кластеров L5@Al39, а также суммарные спиновые плотности на внутреннем сердечнике ρ(L5) и алюминиевом каркасе ρ(Al39). Из табл. 3 видно, что в зависимости от положения атома L в 3d-ряду сердечники L5 претерпевают деформации со значительными изменениями валентных углов и расстояний R(LL) (по сравнению с параметрами свободных кластеров L5, рассчитанных нами в том же приближении). В частности, при изменении L вдоль периода аксиальное расстояние R(La, La) удлиняется от ∼4.8 у Ti5 до ∼5.2 Å у Cr5 и далее монотонно укорачивается до ∼4.1 Å у Cu5, вновь увеличиваясь до ∼4.4 Å у Zn5.
Подобно L2Al42, для кластеров L5@Al39 с атомами L, стоящими в начале и в конце 3d-периода, предпочтительны низкоспиновые состояния (синглет для L5 = Sc5 и Cu5 и дублет для L5 = Ni5). Для кластеров с атомами L в середине периода наиболее выгодны высокоспиновые состояния. У последних соединений (с L в середине периода) при последовательном уменьшении мультиплетности кластера от Smax к Smax −2, Smax −4 и т.д. спиновая плотность сердечников ρ(L5) меняется сравнительно слабо. Так, при изменении мультиплетности кластера Cr5@Al39 в интервале S = = 12−16 плотность ρ(Cr5) сохраняется на уровне 17.0−17.7 е. У кластера Fe5@Al39 при уменьшении S от16 до 8 плотность ρ(Fe5) остается в интервале 13.6−14.3 е. Аналогичная картина наблюдается для ρ(Со5), и в рамках приближенной модели можно полагать, что в этих мультиплетах у атомов Cr, Fe и Co в сердечниках L5 сохраняется по 4, 3 и 2 неспаренных электрона с параллельными спинами, а атомы Ni находятся в нульвалентном состоянии. У кластеров с допантами Ti5 и V5 имеются группы из нескольких низколежаших и близких по энергии мультиплетов (табл. 3).
Особый интерес представляет самый высокоспиновый кластер Mn5@Al39, у которого в интервале S = 17−21 плотность ρ(Mn5) сохраняется в пределах ∼20−21.6 е, что соответствует числу неспаренных электронов у каждого атома Mn, промежуточному между 4 и 5. Ступенчатое уменьшение суммарного спина кластера Mn5@Al39 в интервале S = 17−21 определяется соответствующим увеличением плотности на каркасе ρ(Al39) с противоположным знаком (антиферромагнитный эффект). Кластер Mn5@Al39 может рассматриваться как простейший молекулярный магнит с высоким магнитным моментом и быть полезным при моделировании родственных наноразмерных частиц с более крупными магнитными сердечниками и более высокими магнитными моментами.
Отметим, что, согласно расчетам, у кластеров L5@Al39 с допантами атомов 4d-элементов наиболее выгодными оказываются низкоспиновые состояния и возбуждение уровней с более высокой мультиплетностью связано с затратами энергии. Аналогичная картина отмечалась выше для кластеров Zr2Al42 и Pd2Al44 (табл. 1), где энергии возбуждения триплета и квинтета из синглета лежат в умеренных границах ∼5−12 ккал/моль, а высокоспиновые термы явно невыгодны.
Можно полагать, что результаты расчетов и выводы, полученные для систем L2Al42 и L5Al39, могут быть полезными при распространении DFT-подходов на более крупные наноразмерные кластеры c более высоким содержанием допантов.
Список литературы
Jimmenes-Iszal E., Moreno D., Mercero J.M. et al. // J. Phys. Chem. A. 2014. V. 118. P. 4309.
Zheng M.M., Li S.J., Su Y., Kawazoe Y. // J. Phys. Chem. A. 2013. V. 117. P. 25077.
Costanzo E., van Hemert M.C., Kroes G-J. // J. Phys. Chem. C. 2014. V. 118. P. 513.
Smith J.C., Reber A.C., Khana S.N., Castleman J. // J. Phys. Chem. A. 2014. V. 118. P. 8485.
Das S., Pal S., Krishnamurty S. // J. Phys. Chem. C. 2014. V. 118. P. 19869.
Varano A., Henry D.J., Yarovsky I. // J. Phys. Chem. C. 2014. V. 118. P. 19865.
Brodova I.G., Shirinkina I.G., Petrova A.N. // Lett. Mat. 2011. V. 1. № 1. P. 32.
Akutsu M., Koyashi K., Atobe J. et al. // J. Phys. Chem. A. 2007. V. 110. № 7. P. 12073. doi 10.1021/jp065161p
Charkin O.P., Klimenko N.M. // Russ. J. Inorg. Chem. 2015. V. 60. № 5. P. 577. doi 10.1134/S0036023615050034 [Чаркин О.П., Клименко Н.М. // Журн. неорган. химии. 2015. Т. 60. С. 644.]
Charkin O.P., Klimenko N.M. // Russ. J. Inorg. Chem. 2018. V. 63. № 12. P. 1578. doi 10.1134/S00360-23618120069 [Чаркин О.П., Клименко Н.М. // Журн. неорган. химии. 2018. Т. 63. № 12. С. 1561.]
Charkin O.P., Klimenko N.M., Charkin D.O., Mebel A.M. // Faraday Discuss. Chem. Soc. 2003. V. 124. P. 215.
Frisch M.J. et al. GAUSSIAN-03. Revision B.03. Gaussian, Inc., Pittsburg, PA. 2003.
Becke A.D.J. // J. Chem. Phys. 1993. V. 98. P. 5648.
Lee C., Yang W., Parr R.G. // Phys. Rev. B. 1998. V. 37. P. 785.
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии