

Журнал неорганической химии, 2019, T. 64, № 1, стр. 86-92
Энергии смешения (параметры взаимодействия), пределы замещений и фазовая стабильность твердых растворов Lu1 – xLnxVO4 (Ln = Ce–Yb, Sc,Y)Е. И. Гетьман, С. В. Радио, Л. Б. Игнатова, Л. И. Арданова
Е. И. Гетьман 1, С. В. Радио 1, *, Л. Б. Игнатова 1, Л. И. Арданова 2
1 Донецкий национальный университет имени Васыля Стуса
21021 Винница, ул. 600-летия, 21, Украина
2 Университет штата Миннесота
56001 Манкато, Форд холл, 241, США
* E-mail: radio@donnu.edu.ua
Поступила в редакцию 15.10.2017
После доработки 04.07.2018
Принята к публикации 12.04.2018
Аннотация
В рамках кристаллоэнергетической теории изоморфной смесимости выполнен расчет энергий смешения (параметров взаимодействия) и температурных интервалов стабильности твердых растворов со структурой циркона состава Lu1 – xLnxVO4, где Ln – редкоземельные элементы (РЗЭ), скандий и иттрий. Показано, что с увеличением номера РЗЭ в ряду Ce–Lu вычисленные энергии смешения и критические температуры распада твердых растворов закономерно уменьшаются. Определены пределы замещений при разных температурах, термодинамическая фазовая стабильность и температура перехода в метастабильное состояние. Приведена диаграмма термодинамической стабильности твердых растворов для ванадатов РЗЭ состава Lu1 – xLnxVO4. Результаты работы могут быть использованы при поиске составов матриц и активаторов новых лазерных и других материалов.
ВВЕДЕНИЕ
Твердые растворы ортованадатов редкоземельных элементов (РЗЭ) применяются в качестве материалов для матриц и активаторов оптических квантовых генераторов [1], люминесцентных материалов, сцинтилляторов, оптических поляризаторов, светодиодов [2, 3]. В качестве модификаторов наиболее часто выступают Nd, Dy, Ho, Er, Tm и Yb [4].
Однако физико-химические основы для синтеза твердых растворов и выращивания их монокристаллов – диаграммы состояния и, в частности, области растворимости на основе компонентов этих систем – изучены недостаточно. Это вынуждает исследователей, изучающих лазерные свойства ортованадатов РЗЭ, выбирать состав матриц и модифицирующих добавок либо по аналогии с родственными системами, либо методом “проб и ошибок”, что может привести к излишнему расходу дорогостоящих реактивов и увеличению продолжительности исследований. В качестве примера можно привести информацию из [1], в которой сообщается о попытке выращивания модифицированных неодимом (1 ат. %) монокристаллов методом Чохральского из шихт трех составов, содержащих 10, 20 и 30 ат. % ScVO4 и соответственно 90, 80 и 70 ат. % YVO4. В результате во всех трех случаях получены монокристаллы, содержащие только ~1 ат. % Sc.
Кроме того, не всегда учитывается, что твердые растворы ванадатов, синтезируемые при высоких температурах, склонны к распаду при охлаждении и эксплуатации и могут неконтролируемо изменять фазовый состав и свойства. Это может привести к деградации материалов на их основе.
В настоящее время наибольший интерес исследователей, занимающихся материалами для твердотельных лазеров, вызывают ортованадаты РЗЭ. Они стали альтернативой иттрий-алюминиевому гранату, так как имеют бóльшую теплопроводность, бóльшую ширину и более высокое эффективное сечение поглощения [4]. Первоначально изучали материалы на основе индивидуальных ортованадатов YVO4, LaVO4 и LuVO4 [5]. Позднее было установлено, что более эффективными являются “смешанные” ортованадаты или, точнее, твердые растворы, содержащие лютеций, например LuxY1 – xVO4 [6], которые ранее были недостаточно изучены, по-видимому, вследствие высокой стоимости лютеция. Возможность образования твердых растворов в широких пределах как между компонентами матриц, так и между матрицей и активаторами обусловлена тем, что ортованадаты РЗЭ (кроме ортованадата лантана) относятся к одному структурному типу – циркона.
В связи с этим цель настоящей работы – прогнозирование энергий смешения, пределов замещения и фазовой стабильности твердых растворов ортованадатов со структурой циркона состава Lu1 – xLnxVO4, где Ln – редкоземельные элементы, а также Sc и Y.
РАСЧЕТНАЯ ЧАСТЬ
Выбор метода расчета энергии смешения и его обоснование
В работе [7] были сопоставлены параметры взаимодействия (энергии смешения) в родственных системах La1 – xLnxPO4 (Ln = Eu, Gd), полученные разными авторами за период 2007–2017 гг. В результате использования методов аb initio (2014), strainenergy (2007), dropcalorimetry (2007), аb initio/strainenergy (2016) и dropsolution (2017) их величины в случае La1 –xEuxPO4 соответственно составили 13.4, 5.2, 14 ± 8; 6.2 и 2.5 ± 2.6 кДж/моль, а в случае La1 –xGdxPO4 – 16.5, 6.77, 24 ± 9; 8.6 и 11.4 ± 3.1 кДж/моль соответственно при средних значениях 8.3 и 13.5 кДж/моль для каждой из систем. Очевидно, что наиболее достоверные результаты дает среднее значение. Однако если для La1 – xEuxPO4 наиболее близка к среднему значению (8.3 кДж/моль) величина, полученная методом ab initio/strainenergy (6.2 кДж/моль), то для La1 – xGdxPO4 наиболее близка к среднему значению (13.5 кДж/моль) величина, полученная методом dropsolution (11.4 кДж/моль), т.е. ни одному из проанализированных методов нельзя отдать предпочтение. Поэтому нами в [8] для расчетов энергии смешения в родственных системах La1 – xLnxPO4 был применен принципиально иной способ расчета, разработанный ранее В.С. Урусовым на основе кристаллоэнергетической теории изоморфной смесимости [9, 10]. С помощью этого метода были получены более близкие к средним величинам энергии смешения для La1 – xEuxPO4 и La1 – xGdxPO4, равные соответственно 9.8 (среднее 8.3 кДж/моль) и 13.9 кДж/моль (среднее 13.5 кДж/моль). Поэтому в настоящей работе использован метод В.С. Урусова, особенностью которого является учет не только энергии деформации при замещении различных по размеру структурных единиц, но и энергии, обусловленной различным характером химической связи в компонентах систем, определяемой по разности их степеней ионности. Ранее этот метод не использовался для расчетов взаимозамещений РЗЭ, так как точность электроотрицательностей РЗЭ по шкале Л. Полинга (±0.05) была недостаточной для расчета различий в степенях ионности химической связи в вышеуказанных и родственных им системах.
Основные формулы метода В.С. Урусова
В случае изоструктурности компонентов систем энтальпия смешения, согласно [9], составляет:
Поскольку относительная разность межатомных расстояний катион–тетраэдрический анион (ΔR/R1) в компонентах систем (табл. 1) <0.1, энергия смешения (Qсм) может быть вычислена в приближении регулярных растворов из выражения:
Некоторые исходные данные и результаты расчетов приведены в табл. 1.
Вычисление температур распада твердых растворов
Как видно из табл. 1, во всех системах величины относительных разностей межатомных расстояний катион–тетраэдрический анион существенно меньше 0.1. Следовательно, согласно [9], график зависимости температур распада твердых растворов от состава будет практически симметричным и пределы растворимости можно рассчитывать по уравнению регулярных растворов Беккера [16]:
С увеличением номера РЗЭ вклады в энергию смешения QR, Qε и их суммы (Qсм) закономерно уменьшаются (табл. 1), что объясняется уменьшением различий в размерах замещающихся структурных единиц и степеней ионности химической связи в компонентах. Следует отметить, что величина вклада Qε достигает 50% от Qсм и учитывается, хотя при различии в степенях ионности химической связи в компонентах <0.05 ее учет, согласно [9], необязателен. Необходимость учета Qε в данном случае обусловлена малой величиной (<0.05) размерного параметра ΔR/R1.
Таблица 1.
К расчету энергий смешения (кДж/моль) и критических температур распада твердых растворов Lu1 – xLnxVO4
| Состав | Ln | R, Å | ΔR/R1 | ЭО | εLn | Δε | QR, кДж/моль | Qε, кДж/моль | Qсм, кДж/моль | Ткр, К |
|---|---|---|---|---|---|---|---|---|---|---|
| Lu1 – xLnxVO4 | Ce | 3.776 | 0.0489 | 1.00 | 0.800 | 0.070 | 29.18 | 29.34 | 58.53 | 3490 |
| Pr | 3.760 | 0.0444 | 1.10 | 0.785 | 0.055 | 24.06 | 18.11 | 42.18 | 2520 | |
| Nd | 3.741 | 0.0392 | 1.12 | 0.780 | 0.050 | 18.75 | 14.97 | 33.72 | 2010 | |
| Pm* | 3.725 | 0.0347 | 1.14 | 0.775 | 0.045 | 14.70 | 12.12 | 26.82 | 1600 | |
| Sm | 3.709 | 0.0303 | 1.16 | 0.770 | 0.040 | 11.20 | 9.58 | 20.79 | 1240 | |
| Eu | 3.696 | 0.0267 | 1.18 | 0.766 | 0.036 | 8.70 | 7.76 | 16.46 | 980 | |
| Gd | 3.685 | 0.0236 | 1.21 | 0.760 | 0.030 | 6.80 | 5.39 | 12.19 | 730 | |
| Tb | 3.668 | 0.0189 | 1.19 | 0.764 | 0.034 | 4.36 | 6.92 | 11.28 | 670 | |
| Dy | 3.652 | 0.0144 | 1.25 | 0.752 | 0.022 | 2.53 | 2.90 | 5.43 | 320 | |
| Ho | 3.640 | 0.0111 | 1.25 | 0.752 | 0.022 | 1.50 | 2.90 | 4.40 | 260 | |
| Er | 3.633 | 0.0092 | 1.26 | 0.750 | 0.020 | 1.03 | 2.39 | 3.42 | 200 | |
| Tm | 3.621 | 0.0058 | 1.29 | 0.744 | 0.014 | 0.41 | 1.17 | 1.58 | 90 | |
| Yb | 3.609 | 0.0025 | 1.32 | 0.738 | 0.008 | 0.08 | 0.38 | 0.46 | 30 | |
| Lu | 3.600 | – | 1.37 | 0.730 | – | – | – | – | – | |
| Lu1 – xLnxVO4** | Sc | 3.502 | 0.028 | 1.415 | 0.721 | 0.005 | 9.57 | 0.15 | 9.60 | 570 |
| Y | 3.643 | 0.012 | 1.340 | 0.734 | 0.018 | 1.76 | 1.94 | 3.69 | 220 |
* Межатомное расстояние для ванадата прометия определено как среднее арифметическое из расстояний ванадатов неодима и самария.
** Для составов Lu1 – xLnxVO4 величины ЭО Lu, Sc и Y составляют 1.431, 1.415 и 1.340 соответственно [15].
Расчет критических температур распада Ткр проводили по уравнению [9]:
Как видно из табл. 1, величины Ткр закономерно уменьшаются с увеличением номера РЗЭ. Энергия смешения и критическая температура распада для системы с иттрием близки к соответствующим величинам для систем с гольмием и эрбием вследствие незначительных отличий радиусов их ионов.
Если известна энергия смешения, можно по уравнению Беккера рассчитать по заданному пределу замещения температуру распада твердого раствора для областей с ограниченной растворимостью компонентов. Ошибка расчета, по данным [9], составляет порядка ±100 K.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Изучение зависимости температур распада от атомного номера РЗЭ
По рассчитанным значениям Тp построены их зависимости от номеров РЗЭ для пределов замещения х = 0.01–0.20 (рис. 1) и приведены критические (максимальные) температуры распада твердых растворов (х = 0.5). Эти зависимости позволяют оценить и термодинамическую стабильность твердых растворов.
Рис. 1.
Зависимости вычисленных температур распада твердых растворов систем Lu1–xLnxVO4 от номера РЗЭ (диаграмма термодинамической фазовой стабильности) для пределов замещения 0.01, 0.02, 0.05, 0.10, 0.20 и 0.50.
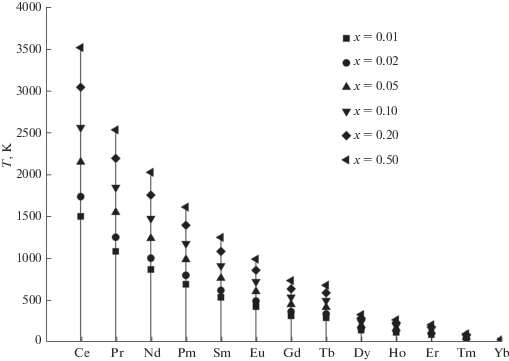
Так, при Т > Ткр (в области выше точек х = 0.5) неограниченные твердые растворы в системах являются термодинамически устойчивыми по всему диапазону концентраций 0 < х < 1. В области ниже точек х = 0.5 при Т < Ткр неограниченные твердые растворы являются термодинамически неустойчивыми и могут распадаться на фазы с ограниченной растворимостью.
По остальным зависимостям можно оценить температуру распада, задав предел замещения, либо оценить предел замещения лютеция на РЗЭ для ограниченных твердых растворов, задав температуру распада. Во втором случае точка пересечения изотермы, проведенной от заданной температуры распада, с вертикалью, проведенной от номера РЗЭ, позволяет определить интервал составов, в котором находится предел замещения, а интерполяция между двумя ближайшими точками гистограммы дает сам предел замещения. Уточнить предел замещения можно, если построить для конкретной системы зависимость вычисленных по уравнению Беккера температур распада от заданного состава.
Известно, что по мере понижения температуры подвижность структурных единиц твердого раствора уменьшается (вследствие уменьшения скорости диффузии) и области растворимости сужаются [9]. Так происходит до тех пор, пока скорость диффузии станет настолько малой, что уменьшение областей растворимости практически прекратится, т.е. произойдет самопроизвольное закаливание и твердые растворы станут метастабильными. Если принять, что температура закаливания близка к минимальной температуре, при которой начинается взаимодействие компонентов в твердой фазе, приводящее к образованию твердого раствора, можно оценить температуру закаливания и область метастабильности в системе.
Оценка области метастабильности твердых растворов ортованадатов РЗЭ
В литературе достаточно много сведений об условиях получения ортованадатов LnVO4 и их твердых растворов ${\text{L}}{{{\text{n}}}_{{1--x}}}{\text{Ln}}_{x}^{'}{\text{V}}{{{\text{O}}}_{4}}$ различными методами, однако в большинстве случаев приводятся промежуточные и конечные температуры синтеза. Но они, по-видимому, могут существенно превышать начальные, так как в меньшей степени зависят от методов синтеза. Так, в частности, при синтезе в твердой фазе [17, 18, 20–22] и методом прокаливания компонентов, полученных из жидкой фазы [19], температура составляет 953–1473 K, при синтезе золь-гель методом [3] и методом магнетронного напыления тонкой пленки [23] – 1273 K.
В случае синтеза из комплексных соединений (NH4)[LnIII(EDTA)] ⋅ xH2O и (NH4)3[VV(O)2(EDTA)] ⋅ ⋅ 0.5H2O с использованием вакуума для удаления растворителя реакции образования ортованадатов РЗЭ и твердых растворов на их основе изучали методом высокотемпературной рентгенографии при ступенчатом повышении температуры через 100 K [24]. Это, по мнению авторов, позволяло получить однородный (с распределением на молекулярном уровне двух комплексов) прекурсор, который обеспечивает понижение температуры взаимодействия по сравнению с другими методами синтеза.
По данным [24], при нагревании прекурсоров для получения YVO4 продукт реакции начинает кристаллизоваться при 773 K, а конечная температура составляет 1073 K (рис. 2).
Рис. 2.
Рентгеновские дифрактограммы, полученные при синтезе YVO4 из прекурсоров, снятые при комнатной температуре (к.т.) после прокаливания при 573 (видны два сигнала эталона), 773, 873, 973 и 1073 К, а также вновь при комнатной температуре после прокаливания при 1073 К [24].
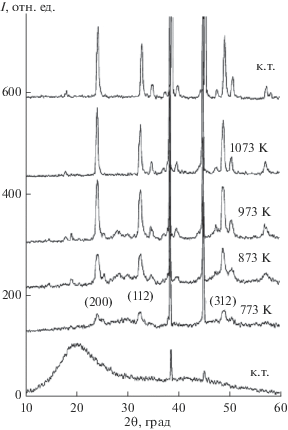
Однако, как видно из рис. 2, при 773 K на рентгенограммах уже видны линии продукта реакции (с индексами (200), (312) и (112)), интенсивность которых увеличивается при дальнейшем повышении температуры. Следовательно, реакция образования и кристаллизации YVO4 начинается не при 773 K, а при меньшей температуре.
По величинам интенсивностей наиболее ярких отражений (200), (112), (312) (рис. 2) построены их зависимости от температуры (рис. 3). После экстраполяции этой зависимости до оси абсцисс определена температура начала реакции и кристаллизации YVO4, равная ~720 K.
Рис. 3.
Зависимость интенсивности отражений (200), (312) и (112) структуры циркона от температуры при синтезе YVO4.
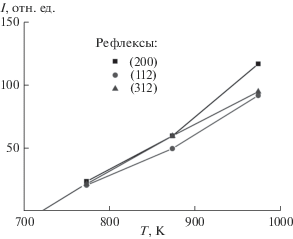
Учитывая близость свойств ортованадатов иттрия и РЗЭ, а также твердых растворов на их основе, можно полагать, что вблизи 720 K скорость диффузии в них такова, что может не только происходить реакция образования YVO4 при подъеме температуры, но и прекращаться распад твердых растворов при их охлаждении ниже 720 K. Поэтому можно предположить, что в системах Lu1 – xLnxVO4 с РЗЭ от Ce до Eu неограниченные твердые растворы, термодинамически стабильные при температурах выше критической (3490–980 K), при понижении температуры в области между Ткр и 720 К становятся термодинамически нестабильными и могут распадаться, образуя ограниченные области. Это происходит, если скорость диффузии и время достаточно велики, чтобы возникли и стали расти устойчивые зародыши новой фазы. При температуре <720 К эти твердые растворы становятся метастабильными.
В системах с РЗЭ от Dy до Yb критические температуры распада (320–30 К) существенно ниже температуры 720 К, неограниченные твердые растворы не должны распадаться при охлаждении и будут метастабильными. Отличие критической температуры для систем с Gd (730 К) и Tb (670 К) от температуры закалки (720 К) не превышает ошибку расчета, поэтому прогнозирование температуры распада неограниченного твердого раствора в этих системах затруднительно.
Сопоставление полученных результатов с литературными данными
Ранее в системах Lu1 – xYxVO4 [6], Lu1 – xScxVO4 [25] и, возможно, в Lu1 − xYbxVO4 [26] (изучена в основном с помощью спектроскопии комбинационного рассеяния) экспериментально установлена неограниченная смесимость компонентов, что согласуется с результатами расчетов, согласно которым критические температуры распада твердых растворов для этих систем составляют соответственно 220, 570 и 30 K (табл. 1). Следовательно, непрерывные ряды твердых растворов в этих системах при температурах выше критических термодинамически стабильны, ниже критических – метастабильны.
Насколько нам известно, в литературе практически нет данных по энергиям смешения и пределам замещения лютеция на РЗЭ для твердых растворов ортованадатов со структурой циркона с ограниченной смесимостью компонентов. Это, конечно, затрудняет оценку достоверности проведенных расчетов. Однако имеются данные по составам и температурам отжига твердых растворов Lu0.99Nd0.01VO4 при 1423 [27] и 1473 K [19] и Lu0.98Nd0.02VO4 при 1273 K [18]. Из графической зависимости вычисленных температур распада твердых растворов Lu1 – xNdxVO4 от мольной доли Nd (рис. 4) следует, что экспериментальные точки, отвечающие твердым растворам состава Lu0.99Nd0.01VO4 (при 1423 и 1473 K) и Lu0.98Nd0.02VO4 (при 1273 K), находятся существенно выше вычисленной кривой распада твердого раствора.
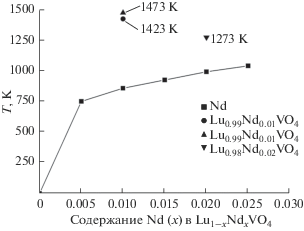
Следовательно, полученные нами результаты расчетов не противоречат экспериментальным данным в том, что твердые растворы, полученные в [18, 19, 27], термодинамически стабильны.
В последнее время появилось много публикаций, например [5, 28, 29], в которых описаны твердые растворы ортованадатов лютеция Lu1 – xLnxVO4, полученные “мягким” гидротермальным синтезом из виннокислых, лимоннокислых или аналогичных растворов при сравнительно низких температурах (от 453 К).
Однако в [30] на примере ряда систем с РЗЭ Tm, Er, Sm, Eu показано, что в действительности при этих температурах образуются образцы, содержащие в своем составе ванадий, частично восстановленный до V4+, карбонильные группы и дефекты кристаллической структуры, которые удаляются при нагревании в интервале температур 523–723 К. Поэтому для завершения синтеза твердых растворов состава Lu1 – xLnxVO4 образцы дополнительно прокаливали при 773 К. Фактически полагали, что при 453 К синтезируется не Lu1 – xLnxVO4, а фаза отличающегося от него состава, хотя и имеющая структуру циркона.
Поэтому нами выполнены расчеты на примере Lu0.95Eu0.05VO4 и Lu0.97Sm0.03VO4, которые показали (рис. 5), что получение твердых растворов таких составов при 453 К действительно невозможно, так как термодинамически стабильными они могут быть при температурах выше 600 и 670 К соответственно, что подтверждается их синтезом при 773 К в [30].
Рис. 5.
Зависимость вычисленных температур распада твердых растворов систем Lu1 – xEuxVO4 и Lu1 – xSmxVO4 от мольной доли РЗЭ, а также экспериментальные температуры двух стадий синтеза Lu0.95Eu0.05VO4 (453 и 773 К) и Lu0.97Sm0.03VO4 (453 и 773 К).
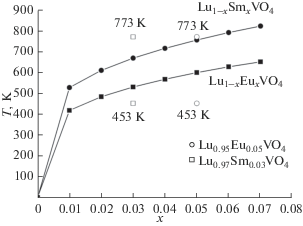
Расчетом также подтверждено, что синтезированные в [30] составы Lu0.95Eu0.05VO4 и Lu0.97Sm0.03VO4 находятся в областях твердых растворов, которые при температуре синтеза (773 К) простираются до х = 0.13 и х = 0.05 соответственно. Таким образом, применение расчетов может быть полезным в тех случаях, когда экспериментальные исследования дают противоречивые результаты.
ЗАКЛЮЧЕНИЕ
В рамках количественной кристаллоэнергетической теории изоморфной смесимости В.С. Урусова проведен расчет энергий смешения и температур распада твердых растворов состава Lu1 – xLnxVO4, образующихся при изоморфном замещении лютеция на РЗЭ, скандий и иттрий в ортованадатах со структурой циркона. Показано, что вклад в энергию смешения составляющих, обусловленных различием в размерах замещающихся структурных единиц и в степенях ионности химической связи, в компонентах практически одинаков. С увеличением номера РЗЭ вычисленные энергии смешения и критические температуры распада твердых растворов закономерно уменьшаются, что обусловлено уменьшением ионных радиусов РЗЭ в ряду от Ce до Lu и различий в степенях ионности химической связи в компонентах систем. Построена диаграмма термодинамической фазовой стабильности твердых растворов для ортованадатов Lu1 – xLnxVO4, позволяющая также прогнозировать пределы замещений лютеция на РЗЭ по заданной температуре либо температуру распада по заданному пределу замещения. Оценена температура перехода твердых растворов в метастабильное состояние. Полученные результаты расчетов не противоречат экспериментальным данным по твердым растворам состава Lu1 – xLnxVO4, описанным в литературе. Результаты работы могут быть использованы при разработке новых лазерных, люминесцентных и других материалов на основе LuVO4, модифицированного редкоземельными элементами.
Список литературы
Zagumennyi A.I., Kutovoi S.A., Sirotkin A.A. et al. // Appl. Phys. B. 2010. V. 99. № 1–2. P. 159.
Чумилина Л.Г. Дис. … канд. хим. наук. Красноярск, 2016. 148 с. www.imet-uran.ru/emp/Chumilina/info.htm (дата доступа: 09.09.2017).
Wei X., Wen J., Li S. et al. // Appl. Phys. Lett. 2014. V. 104. № 16. P. 181904.
Zagumennyi A.I., Popov P.A., Zerouk F. et al. // Quantum Electron+. 2008. V. 38. № 3. P. 227 [Загуменный А.И., Попов П.А., Зерроук Ф. и др. // Квантовая электроника. 2008. Т. 38. № 3. С. 227].
Zhang Y., He H., Zhu W. et al. // Cryst. Eng. Comm. 2011. V. 13. № 21. P. 6471.
Guo L., Wang Zh., Yu H. et al. // AIP Advances. 2011. V. 1. № 4. P. 042143.
Neumeier S., Kegler P., Arinicheva Y. et al. // J. Chem. Thermodyn. 2017. V. 105. P. 396.
Get’man E.I., Radio S.V. // Inorg. Mater. 2017. V. 53. № 7. P. 718 [Гетьман Е.И., Радио С.В. // Неорган. материалы. 2017. Т. 53. № 7. С. 732].
Урусов В.С. Теория изоморфной смесимости. М.: Наука, 1977. 251 с.
Urusov V.S. // Fortschr. Mineral. 1975. V. 52. Special issue. P. 141.
Гетьман Е.И. Изоморфные замещения в вольфраматных и молибдатных системах. Новосибирск: Наука, 1985.147 с.
Бацанов С.С. Структурная химия. Факты и зависимости. М.: Диалог-МГУ, 2000. 292 с.
Xue D., Zuo S., Ratajczak H. // Physica B. 2004. V. 352. № 1–4. P. 99.
Batsanov S.S. // Russ. Chem. Rev. 1968. V. 37. № 5. P. 332 [Бацанов С.С. // Успехи химии. 1968. Т. 37. № 5. С. 778].
Li K., Xue D. // J. Phys. Chem. A. 2006. V. 110. № 39. P. 11332.
Becker R. // Z. Metallkd. 1937. V. 29. P. 245.
Щур С.Л., Левицька К.Є., Неділько С.А. // Наукові праці Донецького національного технічного університету. Сер.: Хімія і хімічна технологія. 2011. № 16. С. 21 (in Ukrainian).
Higuchi M., Shimizu T., Takahashi J. et al. // J. Cryst. Growth. 2005. V. 283. № 1–2. P. 100.
Zhang H., Kong H., Zhao S. et al. // J. Cryst. Growth. 2003. V. 256. № 3–4. P. 292.
Dragomir M. Synthesis and Electronic Properties of Compounds From a Bi2O3–Nd2O3–VOx System. Dissertation (Graduate School, University of Nova Gorica, Nova Gorica, Slovenia) http://www.ung.si/~library/doktorati/fizika/17Dragomir.pdf (accessed on 09 September 2017) 2013. 229 p.
Panchal V., Lopez-Moreno S., Santamaria-Perez D. et al. // Phys. Rev. B. 2011.V. 84. P. 024111.
Cheetham A.K., Sharma N. Novel red phosphors for solid state lighting. U.S. Patent № 2016/0192219. Publ. Date: Aug. 31, 2006.
Minami T., Miyata T., Suzuki Y. et al. // Thin Solid Films. 2004. V. 469–470. P. 65.
Deligne N., Gonze V., Bayot D. et al. // Eur. J. Inorg. Chem. 2008. № 6. P. 896.
Lundgren R.J., Cranswick L.M.D., Bieringer M. // Chem. Mater. 2007. V. 19. № 16. P. 3945.
Levesque Y. Etude optique de materiaux laser: Ybx(Lu,Nd)1 – xVO4. (Faculté des Sciences, Université de Sherbrooke, Sherbrooke. Quebec, Canada) https://www.physique.usherbrooke.ca/pages/sites/default/files/memoireLevesque_Yannick.pdf (accessed on 09 September 2017) 2011. 115 p. (in French)
Zhao S., Zhang H., Wang J. et al. // Opt. Mater. 2016. V. 26. № 3. P. 319.
Gao Y., Feng B., Fang Q. et al. // J. Nanosci. Nanotechnol. 2012. V. 12. № 11. P. 8372.
Preparation of LuVO4 : Eu3+ red fluorescent powder. China Patent CN 101434411 A. Publ. Date: May 20, 2009.
Wang Y., Song Y., Li Y. et al. // New J. Chem. 2017. V. 41. № 2. P. 709.
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии