

Микроэлектроника, 2022, T. 51, № 2, стр. 125-139
DFT моделирование электронной структуры и адсорбция германия в упорядоченном графене с вакансией
М. М. Асадов a, *, С. Н. Мустафаева b, С. С. Гусейнова b, В. Ф. Лукичев c, **
a Институт катализа и неорганической химии им. М.Ф. Hагиева
Национальной академии наук Азербайджана
AZ-1143 Баку, пр. Г. Джавида, Азербайджан
b Институт физики Национальной академии наук Азербайджана
AZ-1143 Баку, пр. Г. Джавида, 131, Азербайджан
c Физико-технологический институт им. К.А. Валиева Российской академии наук
117218 Москва,
Нахимовский просп., 36, корп. 1, Россия
* E-mail: mirasadov@gmail.com
** E-mail: lukichev@ftian.ru
Поступила в редакцию 17.05.2021
После доработки 17.05.2021
Принята к публикации 15.06.2021
- EDN: CVOPEU
- DOI: 10.31857/S0544126922010021
Аннотация
Представлены результаты исследований методом теории функционала плотности (DFT) плотности электронных состояний (DOS) суперъячеек интерфейса дефектного графен/Ge. Изучены закономерности изменения DOS в ряду графен/Ge → графен + вакансия/Ge (GPV). Обсуждаются особенности распределения плотности электронных состояний в интерфейсе упорядоченная вакансия-графен/Ge. На основе DFT-расчетов и физико-химического моделирования изучена природа связи графена с германием и адсорбционные свойства GPV– Ge системы.
1. ВВЕДЕНИЕ
Различные стратегии физико-химической функционализации микро- и наносистем на основе 2D материалов позволяют активизировать их поверхности [1]. Например, за счет функционализации графена происходит сдвиг уровня Ферми, однако степень, до которой может быть настроена запрещенная зона, систематически еще не исследована. Кроме того отсутствуют физико-химические закономерности влияния инертных атомов (адсорбентов) на активацию поверхности графена.
Химическая активность углеродных вакансий (${{V}_{{\text{C}}}}$) в графене к различным атомам [2] может приводить к замещению поверхностных позиций ${{V}_{{\text{C}}}}$. В этом процессе изменяются структурные и электронные свойства графена в зависимости от природы адсорбирующих атомов.
Энергии поверхности графена в различных кристаллографических направлениях заметно отличаются друг от друга. В этом плане интересно также изучение влияния природы и характеристик адсорбата на изменения свойств наноструктуры дефектного графена.
Влияние адсорбции индивидуальных атомов химических элементов с высокой валентностью на свойства графена с ${{V}_{{\text{C}}}}$ недостаточно изучено, тогда как известна масса сведений о допированом полупровдниками дефектном графене. Такие частицы, например, как полупроводниковые кремний и герминий, влияют на электронные и магнитные свойства дефектного графена [3–10]. В связи с этим и возможностями применения графена в наноэлектронике (например, как возможная элементная база графенового полевого транзистора) актуальным является DFT-расчет формируемого спектра поверхностных электронных состояний на границе раздела графен–полупроводник, которая влияет на электронную структуру системы [3].
В настоящей работе приведены результаты DFT-расчетов электронной структуры и адсорбции атомарного германия на физико-химические свойства дефектного графена.
2. МОДЕЛЬ И МЕТОДИКА РАСЧЕТОВ
DFT-расчетные детали. Исследовали теоретическую модель на основе суперъячейки дефектного графена, содержащего 18 атомов углерода и адсорбции атомарного германия (GPV–Ge). Свойства систем на основе монослоя графена вычисляли в рамках периодической теории функционала плотности (DFT [11, 12]) с введением обменно-корреляционного потенциала [12–16].
В DFT задача о многоэлектронном основном состоянии сводится к самосогласованной одноэлектронной форме с помощью уравнения, КШ включающая обменные и корреляционные эффекты [14]. В теории DFT эффекты многоэлектронного обмена и корреляции описываются обменно-корреляционным функционалом электронной плотности $\rho \left( {\vec {r}} \right)$. Этот функционал используется вместо многоэлектронной функции $\psi \left( {\vec {r}} \right)$.
Однако для реальных систем явный вид этого функционала неизвестен, кроме предельного случая, как однородный электронный газ. Поэтому в расчетах DFT в настоящее время используются различные приближения для обменно-корреляционного функционала.
Приближение локальной (спиновой) плотности (LDA) [17], которое ранее использовалось для описания атомной структуры, упругих и колебательных свойств систем, не надежно и может быть стандартом для электронного газа.
В теории DFT энергия изучаемой системы определяется выражением, включающим $\rho \left( {\vec {r}} \right)$ [14]
(1)
$E\left[ {\rho \left( {\vec {r}} \right)} \right] = {{T}_{e}}\left[ {\rho \left( {\vec {r}} \right)} \right] + \smallint {{V}_{0}}\left( {\vec {r}} \right)\rho \left( {\vec {r}} \right)d\vec {r} + \smallint \frac{{\rho \left( {\vec {r}} \right)\rho \left( {\vec {r}{\kern 1pt} '} \right)}}{{\left| {\vec {r} - \vec {r}{\kern 1pt} '} \right|}}d\vec {r}d\vec {r}{\kern 1pt} ' + {{E}_{{{\text{XC}}}}}\left[ {\rho \left( {\vec {r}} \right)} \right]~,$— электронно-ядерное взаимодействие,
— кулоновское взаимодействие между электронами, ${{E}_{{{\text{XC}}}}}\left[ {\rho \left( {\vec {r}} \right)} \right]$ – обменно-корреляционное взаимодействие, которое содержит все остальные электрон-электронные взаимодействия.
Решение выражения (1), позволило записать систему одноэлектронных уравнений [14]:
(2)
$\left\{ { - \frac{1}{2}{{\nabla }^{2}} + {{V}_{{{\text{eff}}}}}\left( {\vec {r}} \right)} \right\}{{\psi }_{i}}\left( {\vec {r}} \right) = {{\varepsilon }_{i}}{{\psi }_{i}}\left( {\vec {r}} \right),$(3)
${{V}_{{{\text{eff}}}}}\left( {\vec {r}} \right) = {{V}_{0}}\left( {\vec {r}} \right) + {{V}_{{\text{C}}}}\left[ {\rho \left( {\vec {r}} \right)} \right] + {{\mu }_{{{\text{XC}}}}}[\rho \left( {\vec {r}} \right).$Здесь ${{V}_{{{\text{eff}}}}}\left( {\vec {r}} \right)$ – эффективный потенциал, в котором движется электрон. В выражении (2) ${{\mu }_{{{\text{XC}}}}}\left[ {\rho \left( {\vec {r}} \right)} \right]$ является функционалом EXC по $\rho \left( {\vec {r}} \right)$, т.е.
(4)
${{\mu }_{{{\text{XC}}}}}\left[ {\rho \left( {\vec {r}} \right)} \right] = \frac{{\delta {{E}_{{{\text{XC}}}}}\left[ {\rho \left( {\vec {r}} \right)} \right]}}{{\delta \rho \left( {\vec {r}} \right)}}.$Обменно-корреляционный функционал строили с применением обобщенного градиентного приближения (generalized gradient approximation – GGA) [18]. Функционалы GGA по сравнению с LDA (local density [14, 18–20]) позволяют лучше описывать свойства систем, например, энергетические барьеры и энергию адсорбции для молекул на поверхности металлов или полупроводников с желаемой точностью [19].
В системах с участием 2D материалов использование приближения GGA по сравнению с ранее предложенной аппроксимацией LDA предпочтительнее. Обменно-корреляционная энергия неоднородной системы в модели LDA локально рассматривается как для электронного газа, т.е. предполагается, что система псевдооднородная. Обменно-корреляционная энергия псевдооднородного электронного газа записывается выражением:
(5)
$E_{{{\text{XC}}}}^{{{\text{LDA}}}}\left[ {\rho \left( {\vec {r}} \right)} \right] = \smallint \rho \left( {\vec {r}} \right){{\varepsilon }_{{{\text{XC}}}}}\left[ {\rho \left( {\vec {r}} \right)} \right]d\vec {r}.$В GGA-PBE (PBE – параметризация Perdew–Burke–Ernzerhof [21]) принимается, что обменно-корреляционная энергия является функцией как электронной плотности (либо спиновой, если система магнитная), так и ее градиента:
(6)
$E_{{{\text{XC}}}}^{{{\text{GGA}}}}\left[ {\rho \left( {\vec {r}} \right)} \right] = \smallint f\left[ {\rho \left( {\vec {r}} \right),\nabla \rho \left( {\vec {r}} \right)} \right]d\vec {r}.$(7)
${{E}_{{{\text{XC}}}}}\left[ {\rho \left( {\vec {r}} \right)} \right] = {{E}_{{\text{X}}}}\left[ {\rho \left( {\vec {r}} \right)} \right] + {{E}_{{\text{C}}}}\left[ {\rho \left( {\vec {r}} \right)} \right].$В рассматриваемой системе GPV–Ge в рамках периодической теории функционала волновые функции вычисляли путем разложения базиса плоских волн. При этом ограничение по энергии составляло 150 Ry, а для плотности заряда – 200 Ry. Элементарную кристаллическую ячейку графена расслабляли и оптимизировали с допуском к силе и напряжению 0.001 и 0.001 эВ/Å3 соответственно. Функционал спин-поляризованной плотности рассчитывали с использованием кода самосогласованного поля плоских волн программным пакетом Atomistix Tool Kit (ATK) [3–5, 22 ].
3. РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Моделирование суперячейки дефектного графена GPV–Ge проводили с оптимизацией геометрии элементарной ячейки графена. Использовали суперячейки, содержащие гексагональную элементарную ячейку графена в плоскости с одной или двумя упорядоченными вакансиями ${{V}_{{\text{C}}}}$ (рис. 1).
Электронные свойства рассчитывали путем вычислений парциальной и полной плотности состояний. Эффективные заряды на адатоме (А) германия и ближайших к нему поверхностных атомах углерода для суперячейки GPV–Ge определяли анализом заселенности по Малликену [23].
Равновесные параметры кристаллической решетки графена и атомные позиции адатома на поверхности GPV–Ge вычисляли с использованием схемы генерации k-точек с плоской сеткой. Расчетный параметр задавали таким образом, чтобы полная энергия суперячейки А–графен с вакансиями воспроизводилась с точностью ≤ 10−3 эВ. Таким образом обеспечивается сходимость результатов DFT-расчетов.
Взаимодействие валентных электронов атомов углерода и адсорбата атома германия с остовным состоянием в GPV–Ge исследовали псевдопотенциальным методом [24–27].
Электронные состояния атомов суперячейки GPV–Ge разделяли на валентные и остовные состояния. Допускали, что остовные состояния локализованы и не участвуют в формировании химических связей. В этом случае псевдопотенциалы остовных состояний исключаются из решения многоэлектронной задачи. В результате вместо ионного потенциала вводится слабый псевдопотенциал, который позволяет описать волновые функции только валентных электронов. Использовали электронные конфигурации атома C – [He] 2s22p2 и адсорбированного атома Ge – [Ar] 3d104s24p2. Здесь [He] и [Ar] относятся к остовным состояниям.
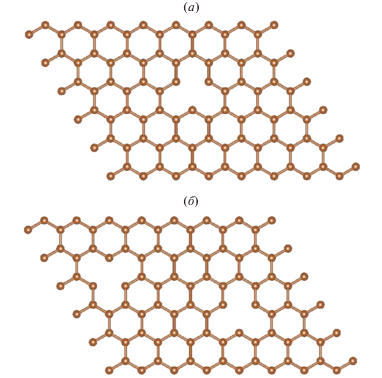
Рис. 1.
Суперъячейки, содержащие гексагональную элементарную ячейку графена с 18 атомами С в плоскости с одной (а) (GPSV) и двумя (б) упорядоченными вакансиями (GPDV).
Атомная структура. Размещение адатома, на поверхности суперячейки GPV–Ge на расстоянии 2 Å от вакансии позволяет “заморозить” систему. За счет этого при вычислениях свойств системы уменьшается сумма всех действующих сил до <0.001 эВ/Å. Атомная структура суперячейки GPV–Ge, а также графена с адатомом германия для двух конфигураций после релаксации системы представлена на рис. 2.
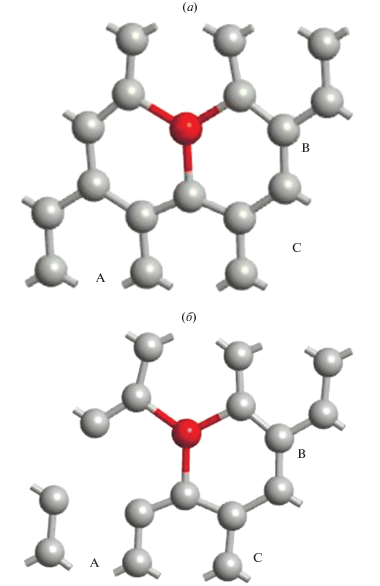
Рис. 2.
Фрагменты положения адатома германия (красный) и разных позиций двух вакансий на поверхности суперъячейки Ge–графен с вакансиями (GPV–Ge): (а) – в изоляции от вакансий; (б) – в непосредственном окружении вакансий.
Стабильность гексагональной кристаллической решетки графена связывается с sp2-орбитальной гибридизацией электронов графена. Комбинация s-орбиталей по координатным осям x, y, z образует три σ-связи и одну π-связь.
Используя структурную схему графена, включающую взаимосвязанные шестиугольники атомов углерода, определили длины связи между ядрами атомов углерода и адатомом на суперъячейки GPV–Ge. Расстояния (Å) между адатомом и заданными атомами углерода на монослое графена вычисляли с учетом эффективных радиусов атомов: ${{r}_{{{\text{ads}}}}} = {{d}_{{{\text{nn}}}}} - {{r}_{{\text{A}}}}$, где ${{d}_{{{\text{nn}}}}}$ – длина связи адсорбата (А), ${{r}_{{\text{A}}}}$– металлический радиус объемного атома A.
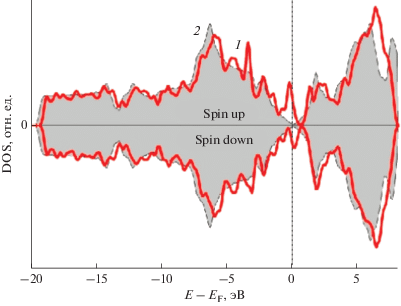
Электронная структура суперъячейки GPV–Ge. Зонная структура и парциальная плотность электронных состояний (PDOS) графена с вакансией характеризуют обменное расщепление состояний на уровне Ферми. На рис. 3 показаны DFT-расчетные электронные спектры атомных конфигураций “чистого графена” и графена с вакансией GPV. Полученные данные DOS согласуются с известными [3, 28].
Рис. 3.
Плотности электронных состояний (DOS) суперъячейки графена, содержащей 18 атомов углерода. 1 – DOS для “чистого” графена; 2 – DOS для графена с моновакансией.
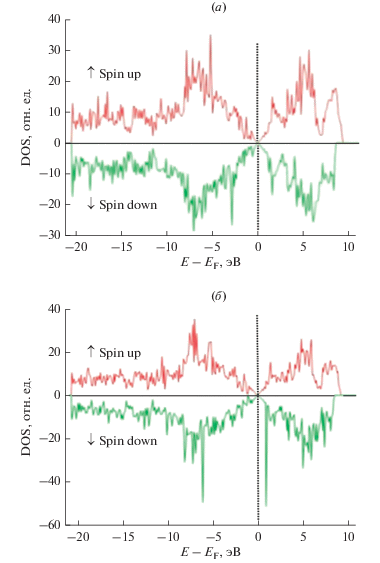
На рис. 4 представлены рассчитанные DOS графена с моновакансией (GPSV) и дивакансией (GPDV). Повышение концентрации вакансий в графене приводит к уменьшению расщепления полосы из-за частичной спиновой поляризации. Ненасыщенные оборванные σ-связи образуют плоские полосы при –0.23, –0.25 и –0.53 эВ при концентрациях вакансий 1, 2 и 3.2% соответственно. Эти σ-полосы показывают большое расщепление по сравнению с π-полосами и соответствуют сильно локализованным состояниям. Плоские полосы способствуют появлению узких пиков в DOS, которые повышают стабильность магнитного упорядочения при высоких температурах [29].
Рис. 4.
Плотности электронных состояний (DOS) суперъячейки графена с моновакансией (а) и дивакансией (б).
Сопоставление спектров DOS конфигурации “чистого” графена и графена с моно- и дивакансией (рис. 3, 4) указывает на незначительную перестройку локальной атомной структуры при упорядочивании вакансий. Расположение вакансий атома углерода в кристаллической решетке графена заметно влияет на РDOS углерода в интервале энергий связи от −17 до 5 эВ.
По данным DFT-расчета зонная структура суперъячейки GPV–Ge соответствует металлическому типу, как для графена. Т.е. заселенность π-состояний на уровне Ферми указывает на металлическую проводимость дефектного графена с адатомом.
Плотность электронных состояний в системе GPV–Ge состоит из вкладов состояний 2p-электронов атомов углерода (C – [He]2s22p2) и незаполненных состояний 3d-электронов атомов Ge (Ge – [Ar]3d104s24p2). Зонные структуры интерфейса двумерной суперячейки графена допированные 4p-элементами Si [3] и Ge, аналогичны.
Парциальные и полные плотности электронных состояний (DOS) суперячейки GPV–Ge приведены на рис. 5 и 6 соответственно. Так как спектры DOS бездефектного (GP–Ge) и дефектного графена (GPV–Ge) аналогичны, то ниже будут рассмотрены данные DFT-расчета суперячейки GPV–Ge (рис. 6).
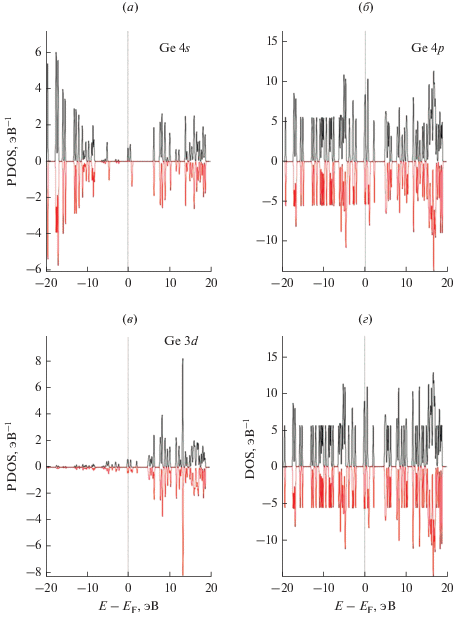
Рис. 5.
Локальные парциальные плотности электронных состояний (PDOS) на один атом германия в связывающих позициях атомов на поверхности суперъячейки графена GP–Ge: (а) – PDOS Ge для s атомной орбитали; (б) – PDOS Ge для p атомной орбитали; (в) – PDOS Ge для d атомной орбитали; (г) – полный DOS системы GP–Ge.
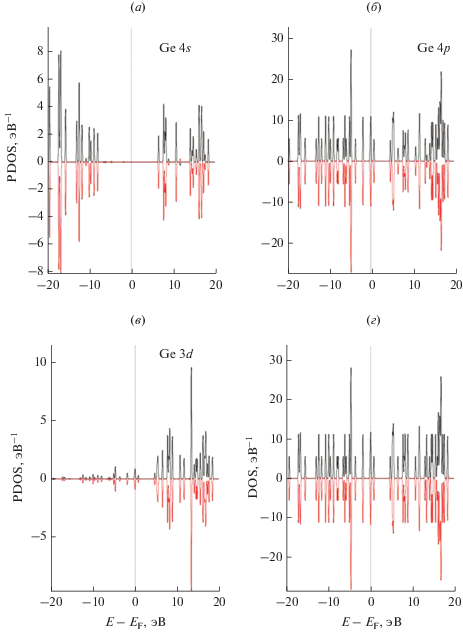
Рис. 6.
Локальные PDOS на один атом германия в связывающих позициях атомов на поверхности суперъячейки моновакансионного графена GPSV–Ge: (а) – PDOS Ge для s атомной орбитали; (б) – PDOS Ge для p атомной орбитали; (в) – PDOS Ge для d атомной орбитали; (г) – полный DOS системы GPV–Ge.
На поверхности монослоев суперъячейки GPV–Ge связи между атомами С и Ge с соответствующими парциальными плотностями электронных состояний заметно отличаются друг от друга.
На спектрах парциальных и полных DOS суперячейки GPV–Ge расчетные энергетические положения пиков позволяют предполагать, что в GPV–Ge за счет физической адсорбции имеет место слабая C2p–Ge 3d-гибридизация между углеродом и адатомом германия.
Состояния 2p-электронов атомов углерода и адатома германия характеризуются в основном двумя пиками в области энергий – 17–16 эВ (рис. 6), т.е. основная часть РDOS атомов германия лежит в указанном интервале энергий. Для указанного интервала энергии связи наблюдается локализация состояний электронов и изменение направленности связи. В этой области энергий отсутствуют РDOS углерода.
В суперъячейке графена связи –C–Ge–C– характеризуются плоскими энергетическими зонами. Эти зоны ответственны за пики РDOS 2p-электронов углерода и адатома Ge. Таким образом, РDOS атомов углерода и адатома германия вносят вклад в полную DOS системы и определяют электронные свойства GPV–Ge.
Межатомные взаимодействия. Рассмотрим физическую адсорбцию в системе поверхность–адсорбат, которая протекает без переноса электронов, т.е. за счет межатомного или межмолекулярного взаимодействия. Расчет потенциальной энергии межмолекулярных взаимодействий молекул с адсорбентами в общем случае представляет трудную задачу. Чем плотнее среда и, следовательно, чем ближе расположены молекулы, тем больше проявляется эффект многочастичного взаимодействия.
Силы, действующие между молекулами, состоят из нескольких составляющих, так что общая сила является их суммой. Энергию отталкивания не удается выразить через характеристики невозмущенных атомов, так как отталкивание проявляется на очень малых расстояниях.
При адсорбции, как известно из термодинамики, уменьшается поверхностная энергия, т.е. уменьшается межфазовая поверхность и поверхностное натяжение. Для вычисления свободной энергии Гельмгольца ($F$) удобно разбивать эту энергию на идеальную часть (${{F}^{{{\text{id}}}}}$) и на поправку к ней (${{F}^{{{\text{ex}}}}}--{\text{избыточная\;часть}}$), которая учитывает различного рода взаимодействия. Т.е. $~F = {{F}^{{{\text{id}}}}} + {{F}^{{{\text{ex}}}}}$. Здесь добавочное слагаемое свободной энергии ${{F}^{{{\text{ex}}}}}$ характеризует взаимодействие частиц, и именно оно определяет физику молекул системы.
Вклад ${{F}^{{{\text{ex}}}}}$ можно представить через функцию, которая выражается через потенциал парного межмолекулярного взаимодействия $v\left( {{{r}_{i}},{{r}_{j}}} \right).$ Однако, его сложно напрямую учесть в выражении для свободной энергии $~F$. Для корректного учета взаимодействия простых частиц, например, через потенциал Леннарда–Джонса (LJ), ${{F}^{{{\text{ex}}}}}$ удобно делить на два слагаемых. Тогда ${{F}^{{{\text{ex}}}}}$ можем записать как: ${{F}^{{{\text{ex}}}}} = {{F}^{{HS}}} + {{F}^{{{\text{att}}}}}$, где ${{F}^{{HS}}}--$ часть энергии, которая связана с короткодействующим отталкиванием, ${{F}^{{{\text{att}}}}}$ – часть энергии, которая связана с дальнодействующим притяжением.
Так как эффект отталкивания проявляется на очень малых расстояниях ($r \approx {{10}^{{ - 8}}}{\text{\;см}}$), то энергия отталкивания должна выражаться через характеристики возмущенных атомов. Здесь $r$ является расстоянием между частицами. Т.е. результат взаимодействия перекрывающихся электронных оболочек системы поверхность–адсорбат приводит к силе отталкивания. При такой силе электроны перекрывающихся оболочек, имеющие параллельные спины, должны переходить на уровни с большей энергией. Квантово-механический расчет энергии отталкивания в системе поверхность–адсорбат сложен, поэтому часто используют различные полуэмпирические способы расчета.
Энергию притяжения в системе также можно делить на несколько составляющих: энергию слабых электромагнитных взаимодействий Ван-дер-Ваальса (они в 10–20 раз слабее, чем силы притяжения между ионами), энергию ориентационного, индукционного и дисперсионного взаимодействия.
В качестве парного потенциала межатомного или межмолекулярного дисперсионного взаимодействия при постоянной температуре для простых систем часто используют вышеуказанный потенциал LJ в виде:
Метод молекулярной динамики, который включает численное решение уравнений движения является эффективным для описания взаимодействия множества частиц. С учетом этого для адсорбционной системы кристалл–адсорбат можно записать ${{f}_{i}} = - \frac{\partial }{{\partial {{r}_{i}}}}U,~\,\,{\text{где\;}}U$ – потенциальная энергия кристалла. Тогда вычисление силы ${{f}_{i}},$ действующей на атомы, будет основываться на потенциальной энергии $U\left( {{{r}^{N}}} \right)$, где ${{r}^{N}} = \left( {{{r}_{1}},{{r}_{2}}, \ldots {{r}_{N}}} \right)$ – полный набор 3N атомных координат. Для простых систем функция $U\left( {{{r}^{N}}} \right)$ ограничивается атомарным (сферическим) описанием. Несферические атомы дополнительно имеют вращательные степени свободы, т.е. вращательные уравнения движения и потенциалы взаимодействия.
Химически несвязанные взаимодействия. В системе поверхность кристалла–адсорбат стабильность структуры кристалла при данной температуре $T$ определяется минимумом свободной энергии: ${{F}_{{{\text{min}}}}}~ = U--TS$, где $S~$ – энтропия. Часть потенциальной энергии ${{U}_{{{\text{non}} - {\text{bonded}}}}}\left( {{{r}^{N}}} \right)$, представляющая не связанные взаимодействия между атомами, разделяется на одно-, двух- и трех-частичное взаимодействие
При периодическом моделировании объемных систем трехчастичное взаимодействие можно опускать. При таких взаимодействиях существенным является в основном парный потенциал $v\left( {{{r}_{i}},{{r}_{j}}} \right) = v\left( {{{r}_{{ij}}}} \right)$. Об экспериментальных и теоретических способах определения этих потенциалов существует обширная литература.
В суперъячейке GPV–Ge примем, что дальнодействующие силы незначительны и притягивающие взаимодействия слабые. В этом случае потенциал взаимодействия частиц может быть усечен. Т.е. в положении минимума потенциала его можно смещать вверх, чтобы получить простую модель взаимодействия тел. DFT-моделирование адсорбции в суперячейке GPV–Ge проводили с учетом того, что атомы на поверхности представляли собой жесткие сферы. В таком случае каждая частица системы имеет объем, недоступный для других атомов. Это позволяет уменьшить влияние объема поверхности, занятого адсорбатом, на деформации структуры (упаковку) углеродных атомов.
При учете точечных электростатических зарядов к модели потенциальной энергии необходимо добавить соответствующий кулоновский потенциал ${{v}^{{{\text{Coulomb}}}}}\left( r \right) = \frac{{{{Q}_{1}}{{Q}_{2}}}}{{4\pi {{\varepsilon }_{0}}r}}$, где ${{Q}_{1}},{{Q}_{2}}$ – заряды, ${{\varepsilon }_{0}}$ – диэлектрическая проницаемость свободного пространства.
Силовые поля для молекулярных систем выражаются как сумма членов парного взаимодействия, которые представляют изменения нескольких вкладов. К ним относятся длины химических связей, валентные углы молекул, вращение атомов вокруг центральной связи, несвязанные электростатические силы и Ван-дер-Ваальса межмолекулярные взаимодействия:
В адсорбционной системе взаимодействия Ван-дер-Ваальса можно описать указанным потенциалом LJ, тогда как электростатические взаимодействия между атомами аппроксимируются законом Кулона с фиксированными точечными зарядами, присущими каждому атому. Такая форма силового поля подходит для моделирования адсорбции как молекулярных, так и немолекулярных систем.
sp2-Углеродные поверхности. Энергии адсорбции и место связывания на поверхности углеродсодержащих небольших молекул (С5, С6), полученные на основе модели, учитывающей молекулярно-механическое силовое поле, сопоставлены в [30] с расчетами DFT. Не поляризуемое силовое поле оказалось адекватным для описания адсорбционных свойств молекул воды на таких углеродсодержащих поверхностях. Однако с увеличением размера sp2-углеродной поверхности, например, фуллерена, результаты моделирования стали неточными, что указывает на влияние поверхностной поляризации.
Расчеты адсорбции. Модифицирование физико-химических свойств графеновых материалов, в частности, связано с изучением теоретических и практических аспектов адсорбции и переноса заряда в системах графен-адсорбированная частица [31]. С этой целью много исследований направлено на функцию поверхности, изучение и оптимизирование важных, например, электрических и сенсорных свойств графена. Однако, известные данные по исследованию механизмов адсорбции и физико-химических свойств на поверхности графена противоречивы и неполные.
При экзотермическом процессе адсорбции, когда адсорбированные частицы (адатом или адсорбат) притягиваются к подложке, энергия высвобождается из системы. Т.е. изменения энтальпии и свободной энергии системы характеризуются отрицательным значением.
Силы взаимодействия адсорбата с адсорбционными центрами приводят к изменению энергии взаимодействия и переносу заряда. Знак этого заряда определяется природой адсорбата. Адсорбаты могут проявлять себя как акцепторами, так и донорами электронной плотности. За счет такого взаимодействия на поверхности измененяется электронная структура материала.
Адсорбция на краевых атомах углерода решетки графена происходит эффективнее из-за наличия оборванных связей [32]. Наоборот бездефектный графен слабо взаимодействует с абсорбатами из-за отсутствия оборванных связей и инертности. Инертность графена обеспечивается за счет влияния сопряженной π-связи в системе графен–адсорбат. Другими словами электрические свойства, сила взаимодействия графена с другими частицами и перенос электронной плотности материалов зависят от наличия структурных дефектов [33], адатомов и функциональных групп.
Смещения положения атомов на поверхности дефектного графена определяли путем составления квантово-химической модели дефектного графена с учетом адсорбции атома германия. Для модели принимали, что графен состоит из sp2-гибридных атомов углерода с атомными орбиталями s, px и py на каждом атоме углерода. Эти орбитали образуют три σ-связи с другими тремя окружающими атомами. Оставшиеся pz-орбитали на каждом атоме углерода перекрываются с соседними атомами углерода и создают заполненную полосу π-орбиталей. Эта полоса состоит из двух составляющих, из валентной зоны и пустой зоны проводимости π*-орбиталей. Разработанную нами модель геометрически оптимизировали и рассчитывали программой jaguar 7.9 по методике [34].
Энергию адсорбции адатома (А) на поверхности графена с вакансией (GPV) рассчитывали выражением [3]:
Рассмотрим процесс адсорбции. В нашей модели химическая адсорбция не будет рассматриваться. Такая адсорбция происходит за счет переноса заряда от адсорбированной частицы к адсорбенту. В этом случае перенос заряда определяется относительным положением плотности состояний на самой высокой занятой молекулярной орбитали и низшей незанятой атомной (или молекулярной) орбитали адсорбата и уровнем Ферми адсорбента.
В нашем случае примем, что адсорбция атома германия происходит физическим путем, т.е. поверхность суперъячейки GPV–Ge химически не активна. В таком случае адсорбат (Ge) прилипает к поверхности за счет слабых межмолекулярных взаимодействий типа Ван-дер-Ваальсовых взаимодействий. Энергии адсорбции таких слабых взаимодействий (Ван-дер-Ваальсово взаимодействие) составляют в среднем ∼10–20 кДж/моль [35], а расстояние между адсорбатом и поверхностью составляет 3–10 Å.
Адсорбция атомарного германия на поверхности монослоя графена оказывает заметное влияние на длину связи между атомами. Адсорбция приводит к перестройке локальной атомной структуры поверхности в окрестности нахождения адсорбата. Длина связи C–C составляет 1.42 Å, а Ge–C (Ван-дер-Ваальсово взаимодействие) – 1.97 Å.
Длина связи (d) атомов ближайшего окружения германия уменьшается от 1.46 для dC–С до 0.11 Å для dC–Ge. С уменьшением d энергия адсорбции адатома $E_{{{\text{ads}}}}^{{{\text{Ge}}}}$ также уменьшается.
При наличии адсорбата Ge и вакансии ${{V}_{{\text{C}}}}$, которые стоят близко друг к другу на поверхности монослоя графена, длина связи C–C увеличивается до ∼0.11 Å. На длину связи C–C кроме адсорбции могут влиять также термодинамические параметры и электронная структура суперъячейки. Таким образом, за счет адсорбции Ge на поверхности моносля дефектного графена и изменения d адатома понижается симметрия кристаллической решетки.
В зависимости от позиции адатома-германия и конфигурации суперъячейки GPV–Ge снижается симметрия решетки графена, что приводит к уменьшению $E_{{{\text{ads}}}}^{{{\text{Ge}}}}$. Это согласуется с литературными данными (табл. 1). Расхождение в значениях $E_{{{\text{ads}}}}^{{{\text{Ge}}}}$ и точность описания характеристик можно связать с выбором потенциала для квантово-химического моделирования и методикой определения поверхности потенциальной энергии (ППЭ), которая характеризуется распределением локальных и глобальных минимумов. На отображениях ППЭ минимумы соответствуют вероятным местам для адсорбции, в то время как седловые точки определяют энергию активации диффузии по разным путям. Адатом Ge помещался в выбранной поверхности на монослое дефектного графена и его поведение спектрально отражалось в виде ППЭ.
Таблица 1.
Длина связи и энергия связи атома германия на позиции моновакансии в суперъячейке графена с 18 атомами С с упорядоченными вакансиями
Увеличение длины связи Ge–C (dC–Ge = 1.97 Å) при адсорбции Ge на поверхности дефектного графена можно связать с большим радиусом Ge по сравнению с С (rGe > rC = 1.22 Å > 0.75 Å), который приводит к снижению энергии адсорбции до $E_{{{\text{ads}}}}^{{{\text{Ge}}}}$ = –0.90 эВ/атом. Таким образом, удельная поверхность, оказывая влияние на количество адсорбционных мест, позволяет усилить взаимодействие абсорбата с поверхностью графена в GPV–Ge. При этом заметно изменяются физико-химические свойства и чувствительность поверхности. По сравнению с Ge, легированным графеном ($E_{{\text{b}}}^{{{\text{Ge}}}}$ = –1.5 эВ/атом [3]), в GPV–Ge наличие адатома Ge приводит к увеличению $E_{{{\text{ads}}}}^{{{\text{Ge}}}}$ и переноса заряда.
Из указанного следует, что термодинамические параметры адсорбции позволяют управлять поверхностными свойствами GPV–Ge. Например, для сенсорного материала, включающего адсорбат, важно оптимизировать энергию адсорбции. Это нужно для обеспечения эффективного связывания адсорбата и хорошей регенерации сенсора путем десорбции.
Адсорбцию германия в GPV–Ge моделировали с учетом выбранных адсорбционных центров, включающих атом углерода с оборванной связью, а также несколько атомов С в ближайшем окружении вакансии. Моно- и дивакансионные дефекты упорядочивали вблизи центра суперъячейки графена. Состояния моновакансии рассмотрели в симметричной конфигурации, которая содержит три оборванные связи на атомах углерода, окружающих одну вакансию.
Зависимость энергии адсорбции $E_{{{\text{ads}}}}^{{{\text{Ge}}}}$ от количества адсорбированных атомов германия ($N$) в GPV–Ge приведена на рис. 7. Величина $E_{{{\text{ads}}}}^{{{\text{Ge}}}}$ характеризует энергию связи ${{E}_{{\text{b}}}}$ и определяет стабильность адсорбата на поверхности графена. Как видно из рис. 7 по отношению к адсорбции германия, моновакансионный дефект более активный, чем дивакансионный. Это согласуется с данными [31] о стабильности моно- и дивакансий на чистой поверхности графена.
Рис. 7.
Зависимость энергии адсорбции атомов германия на поверхности GPV–Ge от количества адсорбата (атомов Ge); 1 – упорядоченная поверхность графенa; 2 – поверхность графенa с моновакансией; 3 – поверхность графенa с дивакансией.
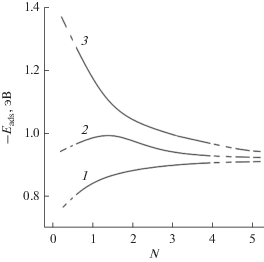
Стабильность атома германия на моновакансионном графене GPSV–Ge (кривая 2) увеличивается на начальном этапе адсорбции при значениях $N$ = 1–2 атомов. Затем зависимость $E_{{{\text{ads}}}}^{{{\text{Ge}}}} = f\left( N \right)$ носит монотонный характер. При значениях $N$ ≥ 2 атомов значение $E_{{{\text{ads}}}}^{{{\text{Ge}}}}$ выходит на плато. В отличие от моновакансионного графена на дивакансионном графене GPDV (кривая 3) значение $E_{{{\text{ads}}}}^{{{\text{Ge}}}}$ германия на поверхности растет на начальных этапах адсорбции вблизи центра двух вакансий. Другими словами значение энергии C–Ge связи (например, при $N$ = 2; ${{E}_{{\text{b}}}}$ = –1.1 эВ) моноатома Ge, адсорбированного на дивакансионном дефекте DV, меньше, чем ${{E}_{{\text{b}}}}$ (–0.96 эВ) для моновакансии SV.
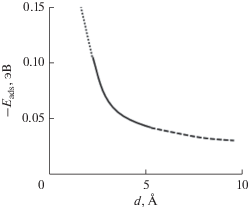
Энергия адсорбции (связи) адатома Ge на поверхности суперъячейки GPSV–Ge вблизи вакансии уменьшается с уменьшением расстояния (d, Å) между адатомом и серединой вакансии (рис. 8). Такое изменение $E_{{{\text{ads}}}}^{{{\text{Ge}}}}$ можно объяснить комбинацией двух эффектов: увеличением деформации вблизи вакансии и уменьшением энергии адсорбции адатома за счет напряженных связей. Все это приводит к эффективному притяжению адатомов дефектами на поверхности GPSV–Ge.
Рис. 8.
Зависимость энергии адсорбции атомов германия около вакансии от расстояния между адатомом Ge и серединой вакансии на поверхности GPV–Ge.
Равновесная концентрация вакансий. Свободная энергия F кристалла, как известно, имеет минимум при заданных температуре T и объеме V в состоянии термодинамического равновесия. В этом состоянии в кристалле всегда будет содержаться равновесное количество точечных дефектов, в частности, вакансий.
Если температура T, давление P и объем V постоянны, то свободная энергия определяется из выражения: F = U – TS, где U – внутренняя энергия кристалла, S – энтропия. Пусть в единице объема кристалла, содержащего $~N~$ атомов, n узлов являются вакансиями и n $ \ll $ N. Тогда минимум свободной энергии F в зависимости от числа вакансий в кристалле определяется условием равновесия: $\frac{{\partial F\left( n \right)}}{{\partial n}} = 0$. При нарушении упорядоченности, в частности, при наличии вакансий в структуре кристалла энтропия возрастает. Вероятность того, что данный узел решетки при температуре Т является вакантным, определяется формулой Больцмана: $P = {\text{exp}}\left( { - E_{f}^{{{\text{V}},p}}{\text{/}}{{k}_{{\text{B}}}}T} \right)$, ${\text{где }}E_{f}^{{{\text{V}},p}}$ − энергия образования вакансии при внешнем давлении ($p$), ${{k}_{{\text{B}}}}$ – постоянная Больцмана. $E_{f}^{{{\text{V}},p}} = E_{f}^{{\text{V}}} + p{{v}_{0}},~{\text{ где }}E_{f}^{{\text{V}}}$ − энергия образования вакансии в отсутствии внешнего давления, ${{v}_{0}}$ – объем вакансии.
Представим кристалл, содержащий $N$ узлов решетки и один точечный дефект. Тогда для $E_{f}^{{\text{V}}}$ можно записать $E_{f}^{{\text{V}}} = E_{f}^{{{\text{SV}}}} - E_{f}^{0}$, где $E_{f}^{{{\text{SV}}}}$ – энергия кристалла с одной вакансией, $E_{f}^{0}$ – энергия этого же бездефектного кристалла. Вакансия формируется путем переноса одного атома из какого-либо узла на поверхность кристалла.
Если дефектный графен состоит из $N$ атомов углерода, то в отсутствии внешнего давления отношение равновесного числа вакансий $n_{{{{{\text{V}}}_{{\text{C}}}}}}^{{{\text{gp}}}}$ к числу занятых атомами мест ($N$ − $n_{{{{{\text{V}}}_{{\text{C}}}}}}^{{{\text{gp}}}}$) при $N$ $ \gg $ $n_{{{{{\text{V}}}_{{\text{C}}}}}}^{{{\text{gp}}}}$ равно равновесной концентрации вакансий (${{C}_{{\text{V}}}})$. Тогда для моновакансии величина ${{C}_{{{\text{SV}}}}} = n_{{{\text{S}}{{{\text{V}}}_{{\text{C}}}}}}^{{{\text{gp}}}}{\text{/}}N = {\text{exp}}\left( { - E_{f}^{{{{{\text{V}}}_{{\text{C}}}}}}{\text{/}}{{k}_{{\text{B}}}}T} \right)$, где $E_{f}^{{{{{\text{V}}}_{{\text{C}}}}}}$ > 0. Концентрации дивакансий с учетом ${{C}_{{{\text{SV}}}}}$ можно определить тажже экспоненциальным уравнением [41]: ${{C}_{{{\text{DV}}}}} = {{\left( {{{C}_{{{\text{SV}}}}}~} \right)}^{2}}\exp \left( { - E_{f}^{{{\text{D}}{{{\text{V}}}_{{\text{C}}}}}}{\text{/}}{{k}_{{\text{B}}}}T} \right)$. Из этого следует, что величина СDV экспоненциально увеличивается при повышении температуры.
На поверхности дефектного графена при Т = = 298 К значение СDV незначительное. С повышением температуры СDV возрастает. Вблизи температуры плавления графена (4510 K [42]) при энергии образования моновакансий $E_{f}^{{{\text{S}}{{{\text{V}}}_{{\text{C}}}}}}$ = 7.3 эВ [5] и дивакансий $E_{f}^{{{\text{D}}{{{\text{V}}}_{{\text{C}}}}}}$ = 7.2 эВ [5] атомов углерода, равновесная концентрация вакансий составляет 7 × 10–9 и 9 × 10–9 соответственно (рис. 9).
Рис. 9.
Зависимость равновесной концентрации вакансий адатома Ge на поверхности суперъячейки GPV–Ge от температуры: 1 – для моновакансии; 2 – для дивакансии.
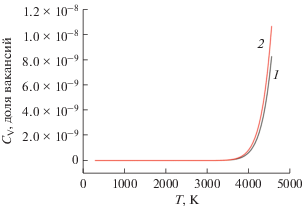
Для сравнения значений концентрации СV вакансий графена с металлами укажем, что для металлов вблизи их точки плавления СV намного больше, например: 2 × 10–4 (для Cu), 9.4 × 10–4 (для Al), 7.2 × 10–4 (для Au), 1.7 × 10–4 (для Ag) [42]. Зависимость CV = f(T) для металлов слабая. Например, в интервале 0 > Т > 1000 К значение моновакансии в меди почти неизменно (∼1.25 эВ [43]).
ЗАКЛЮЧЕНИЕ
В рамках теории функционала плотности полученные равновесные атомные и электронные структуры интерфейса суперячеек графен + моновакансия/Ge(GPSV) → графен + дивакансия/Ge(GPDV) отличаются друг от друга. Это проявляется в распределении спектров плотности электронных состояний (DOS) суперячеек интерфейса дефектный графен/Ge. Указанное изменение происходит за счет физической адсорбции германия на поверхности графена, где имеет место слабая гибридизация между углеродом и адатомом германия: C2p–Ge3d-гибридизация. DOS возможных вариантов позиций вакансий и адатома германия в моносле графена указывает на необходимость выбора подходящего обменно-корреляционного функционала среди известных. Энергетически выгодным является расположение адатома, при котором атомы углерода в GPV располагаются непосредственно вблизи адсорбированного германия. Парциальные DOS атомов углерода и адатома германия вносят вклад в полную DOS суперячейки GPSV и определяют электронные свойства.
Квантово-химическое моделирование GPV–Ge показывает, что изменения длины связи (d) C–C (dC–С = 1.42 Å), С–Ge (dC–Ge = 1.97 Å) и энергии адсорбции ($E_{{{\text{ads}}}}^{{{\text{Ge}}}}$ = –0.9 эВ/атом) адатома германия характеризуются слабым взаимодействием между адатомом и sp2-гибридизированной поверхностью графена за счет сил Ван-дер-Ваальса. Другими словами в выбранной модели GPV–Ge системы при взаимодействии адатома на поверхности графена взаимодействие происходит по механизму физической адсорбции. В этом процессе энергия связи в интерфейсе составляет не более –1 эВ/атом C при равновесном расстоянии между поверхностью германия и атомами углерода dC–Ge = 1.97 Å. В GPV–Ge равновесное расстояние атомов ближайшего окружения германия уменьшается от 1.46 до 1.41 Å для dC–С и от 1.97 до 1.86 Å для dC–Ge. С уменьшением длины связи энергия адсорбции адатома также уменьшается. Сопоставление с известными данными по длине связи C–C, С–Ge и энергии адсорбции адатома германия на позиции моновакансии в суперъячейке GPV–Ge указывает на удовлетворительное согласие результатов в основном по длине связи.
Стабильность адатома германия на моновакансионном графене увеличивается на начальном этапе адсорбции при значениях N = 1–2 атомов (количество адатомов). При значениях N ≥ 2 атомов значение $E_{{{\text{ads}}}}^{{{\text{Ge}}}}$ выходит на плато. На дивакансионном GPDV–Ge графене значение $E_{{{\text{ads}}}}^{{{\text{Ge}}}}$ на поверхности растет на начальных этапах адсорбции вблизи центра двух вакансий. Значение $E_{{{\text{ads}}}}^{{{\text{Ge}}}}$ связи C–Ge c участием дивакансии DV меньше, чем $E_{{{\text{ads}}}}^{{{\text{Ge}}}}$ c участием моновакансии $\left( {E_{{{\text{ads}}}}^{{{\text{Ge}}}}({\text{SV}}) > E_{{{\text{ads}}}}^{{{\text{Ge}}}}({\text{DV}})} \right)$. Величина на поверхности GPV–Ge вблизи вакансии уменьшается с уменьшением расстояния между адатомом и серединой вакансии. Такое уменьшение вблизи вакансии, видимо, связано с влиянием увеличения деформации вблизи вакансии и напряжения связей между атомами.
Настоящая работа выполнена при частичной поддержке Фонда развития науки при Президенте Азербайджанской Республики (проект EİF-BGM-4-RFTFl/2017-21/05/l-M-07) и Российского фонда фундаментальных исследований (проект 18-57-06001 № Az_a 2018).
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Асадов M.M., Мустафаева С.Н., Гусейнова С.C., Лукичев В.Ф. Ab initio расчеты электронных свойств и явления переноса в графеновых материалах // Физика твердого тела. 2020. Т. 62. № 11. С. 1929–1935. [Asadov M.M.,Mustafaeva S.N., Guseinov S.S., Lukichev V.F. Ab Initio Calculations of the Electronic Properties and the Transport Phenomena in Graphene Materials. Physics of the Solid State. 2020. V. 62. № 11. P. 2224–2231. https://doi.org/10.1134/S1063783420110037]
Tsetseris L., Wang B., Pantelides S.T. Substitutional doping of graphene: The role of carbon divacancies. PhysicalReviewB. 2014. V. 89. № 3. P. 035411. https://doi.org/10.1103/PhysRevB.89.035411
Асадов М.М., Гусейнова С.С., Лукичев В.Ф. Ab ınıtıo моделирование электронной и энергетической структуры, а также открытие ширины запрещенной зоны легированного 4p-элементами монослоя графена // Микроэлектроника. 2020. T. 49. № 5. С. 334–343. [Asadov M.M., Guseinov S.S., Lukichev V.F. Ab Initio Modeling of the Electronic and Energy Structure and Opening the Band Gap of a 4p-Element-Doped Graphene Monolayer // Russian Microelectronics. 2020. V. 49. № 5. P. 314–323. https://doi.org/10.1134/S1063739720050030]10.1134/S1063739720050030]https://doi.org/10.31857/S0544126920050038
Лукичев В.Ф., Амиров И.И. Исследования и разработки в области микро- и наносистемной техники // История науки и техники. 2018. № 8. С. 92–99.
Асадов M.M., Мустафаева С.Н., Гусейнова С.C., Лукичев В.Ф., Тагиев Д.Б. Ab initio моделирование влияния расположения и свойств упорядоченных вакансий на магнитное состояние монослоя графена // Физика твердого тела. 2021. Т. 63. № 5. С. 680–689. [Asadov M.M., Mustafaeva S.N., Guseinova S.S., Lukichev V.F., Tagiev D.B. Ab initio modeling of the location and properties of ordered vacancies on the magnetic state of a graphene monolayer // Physics of the Solid State. 2021. V. 63. № 5. P. 680–689.]https://doi.org/10.1134/S1063783421050036
Akturk E., Ataca C., Ciraci S. Effects of silicon and germanium adsorbed on graphene. 2010. V. 96. № 12. P. 123112. https://doi.org/10.1063/1.3368704
Loghavia M.M., Mohammadi-Maneshb H., Eqraa R., Ghasemia A., Babaieea M. DFT Study of Adsorption of Lithium on Si, Ge-doped Divacancy Defected Graphene as Anode Material of Li-ion Battery // Phys. Chem. Res. 2018. V. 6 No. 4. P. 871–878. https://doi.org/10.22036/pcr.2018.148943.1543
Gao H., Zhou J., Lu M., Fa W., Chen Y. First-principles study of the IVA group atoms adsorption on graphene. J. Appl.Phys. 2010. V. 107. № 11. P. 114311. https://doi.org/10.1063/1.3437640
Ould Ne M.L., Abbassi A., El hachimi A.G., Ez-Zahraouy A.B., El Kenz A. Electronic optical, properties and widening band gap of graphene with Ge doping // Opt Quant Electron. 2017. V. 49. P. 218. https://doi.org/10.1007/s11082-017-1024-5
Jules C., Moréac A., Delhaye G., Lépine B., Tricot S., Turban P., Schieffer P., Le Breton J.-C. Reduction of Schottky Barrier Height at Graphene/Germanium Interface with Surface Passivation // Applied Sciences. MDPI. 2019. V. 9. No. 23. P. 5014.https://doi.org/10.3390/app9235014
Hohenberg P., Kohn W. Inhomogeneous electron gas // Phys. Rev. 1964. V. 136. № 3B. P. B864–B871. https://doi.org/10.1103/PhysRev.136.B864
Koch W.A., Holthausen M.C. Chemist’s guide to density functional theory. 2nd edition / Wiley-VCH Verlag GmbH. (2001). 293 p. ISBN: 978-3-527-30372-4.
Fulde P. Electron Correlations in Molecules and Solids. Springer Series in Solid-State Sciences. Springer-Verlag Berlin Heidelberg. 2002. 483 p. https://doi.org/10.1007/978-3-642-57809-0
Kohn W., Sham L.J. Self-consistent equations including exchange and correlation effects // Phys. Rev. 1965. V. 140. № 4A. C. A1133–A1138. https://doi.org/10.1103/physrev.140.a1133
Kohn W., Meir Y., Makarov D.E. van der Waals Energies in Density Functional Theory // Physical Review Letters. 1998. V. 80. № 19. P. 4153–4156. https://doi.org/10.1103/PhysRevLett.80.4153
Parr R.G., Yang W. Density-Functional Theory of Atoms and Molecules Oxford University Press, New York, 1989. 352 p. ISBN: 9780195092769
Jones R.O., Gunnarsson O. The density functional formalism, its applications and prospects // Rev. Mod. Phys. 1989. V. 61. № 3. P. 689–746. https://doi.org/10.1103/RevModPhys.61.689
Ernzerhof M., Perdew J.P., Burke K. Density functionals: Where do they come from, why do they work?. In: Nalewajski R.F. (eds) Density Functional Theory I. Topics in Current Chemistry, vol 180. Springer, Berlin, Heidelberg. 1996. https://doi.org/10.1007/3-540-61091-X_1
Zupan A., Burke K., Ernzerhof M., Perdew J.P. Distributions and averages of electron density parameters: Explaining the effects of gradient corrections // J. Chem. Phys. V. 106. № 24. P. 10184–10193.https://doi.org/10.1063/1.474101
Perdew J.P., Zunger A. Self-interaction correction to density-functional approximations for many-electron systems. Phys. Rev. B. 1981. V. 23. № 10. P. 5048–5079. https://doi.org/10.1103/physrevb.23.5048
Perdew J.P., Burke K., Ernzerhof M. Generalized gradient approximation made simple // Phys. Rev. Lett. 1996. V. 77. № 18. P. 3865–3868. https://doi.org/10.1103/physrevlett.77.3865
Yengejeh S.I., Kazemi S.A., Öchsner A.A. Primer on the Geometry of Carbon Nanotubes and Their Modifications. Springer International Publishing. 2015. 73 p. https://doi.org/10.1007/978-3-319-14986-8_1
Mulliken R.S. Electronic Population Analysis on LCAOMO Molecular Wave Functions // J. Chem. Phys. 1955. V. 23. № 10. P. 1833–1840. https://doi.org/10.1063/1.1740588
Hamann D., Schlüter M., Chiang C. Norm-conserving pseudopotentials // Phys. Rev. Lett. 1979. V. 43. № 20. P. 1494–1497. https://doi.org/10.1103/PhysRevLett.43.1494
Kleinman L., Bylander D.M. Efficacious form for model pseudopotentials // Phys. Rev. Lett. 1982. V. 48. № 20. P. 1425–1428. https://doi.org/10.1103/PhysRevLett.48.1425
Pickett W.E. Pseudopotential methods in condensed matter applications // Comput. Phys. Reports. 1989. V. 9. № 3. P. 115–197. https://doi.org/10.1016/0167-7977(89)90002-6
Vanderbilt D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism // Phys. Rev. B. 1990. V. 41. № 11. P. 7892–7895. https://doi.org/10.1103/PhysRevB.41.7892
Ali M., Pi X., Liu Y., Yang D. Electronic and magnetic properties of graphene, silicene and germanene withvarying vacancy concentration // AIP Advances. 2017. V. 7. № 4. P. 045308-1–11. https://doi.org/10.1063/1.4980836
Edwards D.M., Katsnelson M.I. High-temperature ferromagnetism of sp electrons in narrow impurity bands: Application to CaB6 // Journal of Physics Condensed Matter. 2006. V. 18. № 31. P. 7209–7225. https://doi.org/10.1088/0953-8984/18/31/016
Schlick T. Molecular Modeling and Simulation. Springer Science+Business Media, LLC. New York, 2002. 634 p. https://doi.org/10.1007/978-0-387-22464-0
El-Barbary A.A., Telling R.H., Ewels C.P., Heggie M.I., Briddon P.R. Structure and energetics of the vacancy in graphite // Phys. Rev. B: Condens. Matter. 2003. V. 68. № 14. P. 144107-1–11.https://doi.org/10.1103/PhysRevB.68.144107
Huang B., Li Z.Y., Liu Z.R., Zhou G., Hao S.G., Wu J., Gu B.-L., Duan W. Adsorption of gas molecules on graphene nanoribbons and its implication for nanoscale molecule sensor // J. Phys. Chem. C. 2008. V. 112. P.13442–13446. https://doi.org/10.1021/jp8021024
Zhang Y.-H., Chen Y.-B., Zhou K.-G., Liu C.-H., Zeng J., Zhang H.-L., Peng Y. Improving gas sensing properties of graphene by introducing dopants and defects: a first-principles study // Nanotechnology. 2009. V. 20. № 18. P. 185504. https://doi.org/10.1088/0957-4484/20/18/185504
Jaguar 7.9. User Manual. Schrödinger, LLC, New York, NY, 2012. 368 p.
Israelchvili J.N. Intermolecular and Surface Forces, Third Edition. Academic Press, Elsevier. Amsterdam. 2011. 706 p. ISBN 9780123919274.
Gao H., Zhou J., Lu M., Fa W., Chen Y. First-principles study of the IVA group atoms adsorption on graphene // J. Appl.Phys. 2010. V. 107. № 11. P. 114311-1–6. https://doi.org/10.1063/1.3437640
Chen D.M., Shenai P.M., Zhao Y. Tight binding description on band gap opening of pyrenedispersed graphene // Phys. Chem. Chem. Phys. 2011. V. 13. P. 1515–1520. https://doi.org/10.1039/C0CP00909A
Rafique M.,Yong S., Ali I., Ahmed I. Germanium Atom Substitution in Monolayer Graphene: A First-principles Study // IOP Conf. Ser.: Mater. Sci. Eng. 2018. V. 422. P. 012010. https://doi.org/10.1088/1757-899X/422/1/012010
Akturk E., Ataca C., Ciraci S. Effects of silicon and germanium adsorbed on graphene // Appl. Phys. Lett. 2010. V. 96. № 12. P. 123112-1–3. https://doi.org/10.1063/1.3368704
Ould Ne M.L., Abbassi A., El hachimi A.G., Benyoussef A., Ez-Zahraouy H., El Kenz A. Electronic optical, properties and widening band gap of graphene with Ge doping // Opt Quant Electron. 2017. V. 49. P. 218-1–13. https://doi.org/10.1007/s11082-017-1024-5
Трушин Ю.В. Физические основы материаловедения. Издание второе (переработанное и дополненное). СПб.: Изд-во Академ. ун-та, 2015. 356 с. ISBN 978-5-9064-3304-642.
Zakharchenko K.V., Fasolino A., Los J.H., Katsnelson M.I. Melting of graphene: from two to one dimension // J. Phys.: Condens. Matter. 2011. V. 23. № 20. P. 202202. https://doi.org/10.1088/0953-8984/23/20/202202
Новиков И.И. Дефекты кристаллического строения металлов. М: Металлургия, 1983. 232 с.
Zhao L., Najafabadi R., Srolovitz D.J. Finite temperature vacancy formation thermodynamics: local harmonic and quasiharmonic studies // Modelling Simul. Mater. Sci. Eng. 1993. V. 1. № 4. P. 539–551. https://doi.org/10.1088/0965-0393/1/4/015
Дополнительные материалы отсутствуют.
Инструменты
Микроэлектроника