

Известия РАН. Механика твердого тела, 2022, № 6, стр. 79-96
ВЛИЯНИЕ КРИСТАЛЛИЧЕСКОЙ СТРУКТУРЫ И ТИПА МЕЖАТОМНОЙ СВЯЗИ НА УПРУГИЕ СВОЙСТВА ОДНОАТОМНЫХ И ДВУХАТОМНЫХ КУБИЧЕСКИХ КРИСТАЛЛОВ
А. И. Епишин a, *, Д. С. Лисовенко b, **
a Институт структурной макрокинетики и проблем материаловедения им. А.Г. Мержанова РАН
Черноголовка, Россия
b Институт проблем механики им. А.Ю. Ишлинского РАН
Москва, Россия
* E-mail: a.epishin2021@gmail.com
** E-mail: lisovenk@ipmnet.ru
Поступила в редакцию 01.06.2022
После доработки 24.06.2022
Принята к публикации 27.06.2022
- EDN: IUAJGI
- DOI: 10.31857/S0572329922060058
Аннотация
Проанализирована зависимость упругих свойств одноатомных (простых веществ) и двухатомных (соединений типа АВ) кристаллов кубической сингонии от типов их кристаллической структуры и межатомной связи. Показано, что упругие свойства этих кристаллов следуют одному из двух трендов – ковалентному или ионному. Продемонстрировано, что ауксетики (материалы с отрицательным коэффициентом Пуассона) выявляются среди одноатомных кристаллов со структурой А1 (ГЦК), относящимся к щелочноземельным, переходным и постпереходным металлам, актиноидам и лантаноидам, и одноатомных кристаллов со структурой А2 (ОЦК), относящимся к щелочным металлам. Среди бинарных двухатомных соединений наибольшее число ауксетиков выявляется среди алмазоподобных кристаллов со структурой цинковой обманки B3, имеющих стехиометрию ANB8–N.
1. Введение. Изучение упругих свойств материалов является постоянно актуальной задачей, так как величины упругих характеристик требуются во множестве инженерных и научных расчетов. Значительная часть твердых тел имеет кубическую кристаллическую структуру, в том числе кристаллы многих простых веществ и двухатомных бинарных соединений со стехиометрией АВ. Кубические кристаллы многих металлов имеют гранецентрированную структуру А1 (ГЦК структура меди) или объемно-центрированную структуру А2 (ОЦК структура вольфрама), элементы подгруппы IVA (C, Si, Ge, α-Sn) имеют структуру алмаза А4, а Po и U – примитивную кубическую структуру Ah. Бинарные соединения АВ образуют следующие кубические структуры: В1 – структуру каменной соли NaCl, состоящую из двух ГЦК подрешеток элементов А и В, с расположением атомов в единой решетке как в примитивной решетке; В2 – структуру хлорида цезия CsCl, состоящую из двух примитивных подрешеток А и В, с расположением атомов в единой решетке как в ОЦК; или В3 – структуру цинковой обманки β‑ZnS, также состоящей из двух ГЦК – подрешеток, в которой, однако, А- и В-подрешетки расположены относительно друг друга иначе, чем в структуре NaCl – ближайшими соседями каждого атома являются четыре атома противоположного типа, расположенные в вершинах правильного тетраэдра, т.е. также как в структуре алмаза.
Упругие свойства кристаллов с кубической структурой исследовали во многих работах [1–21]. В [1–3] анализировались стационарные и экстремальные значения модуля Юнга. В этих работах было продемонстрировано, что кристаллы с кубической структурой имеют три стационарных значения модуля Юнга. Два из трех значений могут являться экстремальными значениями. В [4] были проанализированы кристаллографические направления, в которых могут наблюдаться экстремальные значения модуля сдвига кубических кристаллов. Авторами работы [5] была продемонстрирована связь между экстремальными значениями модуля Юнга и модуля сдвига. В [6–21] был проведен анализ изменчивости коэффициента Пуассона для кристаллов с кубической структурой. В теории упругости анизотропного тела положительная определенность энергии деформации не накладывает каких-либо общих ограничений на коэффициенты Пуассона упругих материалов как в случае кристаллов с кубической структурой, так и кристаллов с триклинной, моноклинной, орторомбической, тетрагональной, тригональной (ромбоэдрической) и гексагональной структурой [6]. В [7–21] было продемонстрировано, что кубические кристаллы могут принимать не только положительные значения коэффициента Пуассона, но и отрицательные значения. Материалы с отрицательным коэффициентом Пуассона стали называть ауксетиками. Термин “ауксетики” был предложен профессором Кеном Эвансом [22] для материалов, которые расширяются в поперечном направлении при приложении к ним растягивающей продольной нагрузки. Все кристаллические материалы делятся на неауксетики (материалы, у которых коэффициент Пуассона принимает положительные значения при любой ориентации кристаллической решетки относительно направления растяжения), частичные ауксетики (материалы, у которых коэффициент Пуассона принимает как положительные, так и отрицательные значения в зависимости от ориентации кристаллической решетки относительно направления растяжения) и полные ауксетики (материалы, у которых коэффициент Пуассона принимает отрицательные значения при любой ориентации кристаллической решетки относительно направления растяжения) [14, 15, 18]. В [15, 18] показано, что более чем для 300 кристаллов коэффициент Пуассона может принимать отрицательные значения. Частичными ауксетиками являются кристаллы таких металлов как железо, никель, золото, серебро, литий, натрий, калий, кальций и т.д. В [12, 16] проанализированы экстремальные значения коэффициента Пуассона для кристаллов с кубической структурой. В этих работах показано, что кроме четырех основных стационарных значений, могут наблюдаться экстремально высокие значения коэффициента при растяжении в направлении близком к [111]. Такие значения коэффициента Пуассона свойственны метастабильным кристаллам, таким как кристаллы с эффектом памяти формы [16]. В [10] для ОЦК-кристаллов была предложена структурная модель, поясняющая ауксетическое поведение кристаллов. В [18] представлен обзор ауксетических материалов с кубической структурой, таких как кубические кристаллы, слоистые композиты и нано/микротрубки.
В настоящей работе будут рассматриваться одноатомные (простые вещества) и двухатомные (соединений типа АВ) кристаллы вышеуказанных структур кубической сингонии с металлической, ионной и ковалентной связью. Задачей данного исследования является установление зависимостей упругих свойств кристаллов от типа их структуры и характера межатомной связи. При анализе будет использоваться характеристическая диаграмма упругих свойств, предложенная авторами ранее [16]. Особое внимание в исследовании будет уделено структурным и физическим факторам, обуславливающим ауксетические упругие свойства кристаллов. В последнее время этим материалам уделяется большое внимание [8, 18, 23–26] потому, что это свойство материалов-ауксетиков интересно с практической точки зрения, так как оно может быть использовано при конструировании специальных устройств, а также представляет теоретический интерес, так как исходя из такого аномального упругого поведения твердых тел можно судить о природе межатомного взаимодействия.
2. Характеристические диаграммы упругих свойств кубических кристаллов. Перед тем как перейти к рассмотрению диаграмм упругих свойств кубических кристаллов определимся с обозначением используемых переменных. Упругие свойства кубических кристаллов могут быть охарактеризованы либо тремя компонентами упругих жесткостей ${{c}_{{11}}}$, ${{c}_{{12}}}$, ${{c}_{{44}}}$ либо тремя компонентами упругих податливостей ${{s}_{{11}}}$, ${{s}_{{12}}}$, ${{s}_{{44}}}$, которые связаны между собой соотношениями
где $s{\text{*}} = s_{{11}}^{2} + {{s}_{{11}}}{{s}_{{12}}} - 2s_{{12}}^{2} = \left( {{{s}_{{11}}} - {{s}_{{12}}}} \right)\left( {{{s}_{{11}}} + 2{{s}_{{12}}}} \right)$. На компоненты жесткости накладываются следующие термодинамические ограничения: ${{c}_{{11}}} > 0$, $ - 0.5 < {{c}_{{12}}}{\text{/}}{{c}_{{11}}} < 1~$ и ${{c}_{{44}}} > 0$. При этом соотношения (2.1), (2.2) и (2.3) инвариантны относительно взаимной замены переменных ${{c}_{{ij}}} \leftrightarrow {{s}_{{ij}}}$.Упругую анизотропию кубических кристаллов будем описывать коэффициентом анизотропии A, равного комбинации упругих податливостей $s = {{s}_{{11}}} - {{s}_{{12}}} - {{s}_{{44}}}{\text{/}}2$, нормализованной на ${{s}_{{11}}}$
Для кубических кристаллов преобразование компонент тензора упругих податливостей ${{s}_{{ijkl}}}$ при изменении системы координат имеет следующий вид
где ${{s}_{{ijkl}}}$ и ${{s}_{{i'j'k'l'}}}$ – соответственно компоненты тензора упругих податливостей в базовой системе координат 〈001〉 и произвольной новой системе координат, ${{{{\Phi }}}_{{i'j'k'l'}}}$ = = $2\mathop \sum \limits_{m = 1}^3 {{l}_{{mi'}}}{{l}_{{mj'}}}{{l}_{{mk'}}}{{l}_{{ml'}}}$ – ориентационный фактор, и ${{l}_{{mi'}}}$ – направляющие косинусы между m-й и i´-й осями старой и новой систем координат. Из (2.5) видно, что кубические кристаллы упруго изотропны, т.е. ${{s}_{{i'j'k'l'}}}$ не зависит от ориентационного фактора ${{{{\Phi }}}_{{i'j'k'l'}}}$, если $s$ = 0. Из соотношения (2.5) следуют соотношения для модулей нормальной упругости ${{E}_{{\left[ {hkl} \right]}}}$Индексы в обозначениях модуля Юнга указывают направления растяжения. В случае положительной анизотропии (A > 0) выполняется неравенство ${{E}_{{\left[ {111} \right]}}} > {{E}_{{\left[ {011} \right]}}} > {{E}_{{\left[ {001} \right]}}}$, тогда как при отрицательной анизотропии (A < 0) имеет место обратная закономерность ${{E}_{{\left[ {111} \right]}}} < {{E}_{{\left[ {011} \right]}}} < {{E}_{{\left[ {001} \right]}}}$. Следует отметить, что характеризация упругой анизотропии коэффициентом $A$ подобна характеризации известным коэффициентом Зенера ${{A}^{Z}} = 2{{c}_{{44}}}{\text{/}}\left( {{{c}_{{11}}} - {{c}_{{12}}}} \right) = 2\left( {{{s}_{{11}}} - {{s}_{{12}}}} \right){\text{/}}{{s}_{{44}}}$, поскольку они связаны между собой соотношением $A = \left( {{{s}_{{44}}}{\text{/}}2{{s}_{{11}}}} \right)({{A}^{Z}} - 1)$, где ${{s}_{{44}}}{\text{/}}2{{s}_{{11}}} > 0$. Поэтому случаи отрицательной, нулевой или положительной анизотропии, характеризуемые коэффициентом A, т.е. случаи $A > 0,{\text{\;}}A = 0,{\text{\;}}A < 0$, соответствуют случаям ${{A}^{Z}} > 1,{\text{\;}}{{A}^{Z}} = 1$, AZ < 1.
Вторым параметром упругой анизотропии, который будет анализироваться в настоящей работе, является коэффициент Пуассона в базовой системе координат 〈001〉
(2.8)
${{\nu }_{0}} \equiv {{\nu }_{{\left[ {001} \right]\left[ {010} \right]}}} = - {{s}_{{12}}}{\text{/}}{{s}_{{11}}}$Здесь и далее индексы во второй квадратной скобке указывают направления растяжения, а индексы в первой квадратной скобке – направление поперечной деформации. Использование параметров A и ν0 в данной работе объясняется тем, что экстремальные значения коэффициента Пуассона могут быть описаны ими через простые соотношения
(2.9)
${{\nu }_{{\left[ {01\bar {1}} \right]\left[ {011} \right]}}} = \left( {{{\nu }_{0}} - A{\text{/}}2} \right){\text{/}}\left( {1 - A{\text{/}}2} \right)$(2.10)
${{\nu }_{{\left[ {100} \right]\left[ {011} \right]}}} = {{\nu }_{0}}{\text{/}}\left( {1 - A{\text{/}}2} \right)$На рис. 1 представлены двумерные диаграммы упругих свойств кубических кристаллов. Первая (рис. 1,а), с координатными осями ${{c}_{{44}}}{\text{/}}{{c}_{{11}}}$ и ${{c}_{{12}}}{\text{/}}{{c}_{{11}}}$, была предложена Блэкманом в [27], вторая (рис. 1,b), с осями A и ν0, – Епишиным и Лисовенко в [16]. Жирные черные линии на диаграммах – границы термодинамически допустимых значений упругих констант. Штриховые линии – границы характеристических областей коэффициента Пуассона: 1-я область – “Неауксетики” (Nonauxetics), 2-я и 3-я области – “Частичные ауксетики” (Partial auxetics) и 4-я – “Полные ауксетики” (Complete auxetics). Видно, что на обеих диаграммах нижние и верхние характеристические области разделены горизонтальными прямыми линиями, но негоризонтальные границы имеют разный вид. На диаграмме $A - {{\nu }_{0}}$-негоризонтальная граница представляет собой прямую линию, описываемую простым уравнением:
Рис. 1.
Диаграммы упругих свойств кубических кристаллов. (а) Блэкмана ${{c}_{{44}}}{\text{/}}{{c}_{{11}}} - {{c}_{{12}}}{\text{/}}{{c}_{{11}}}$, (b) Епишина и Лисовенко $A - {{\nu }_{0}}$.
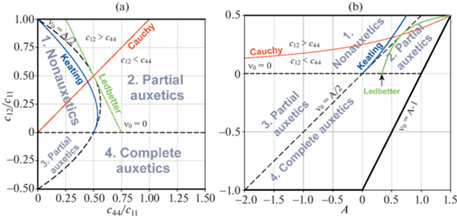
Эта линия начинается на нижней границе допустимых значений ν0, с точки с координатами (A = –2; ν0 = –1), проходит через начало координат (A = 0; ${{\nu }_{0}}$ = 0) и заканчивается на верхней границе допустимых значений ν0 в точке (A = 1; ${{\nu }_{0}}$ = 0.5).
На диаграмме ${{c}_{{44}}}{\text{/}}{{c}_{{11}}} - {{c}_{{12}}}{\text{/}}{{c}_{{11}}}$ негоризонтальная граница описывается квадратичным уравнением
(2.13)
$\frac{{{{c}_{{44}}}}}{{{{c}_{{11}}}}} = \frac{1}{2}\left[ {1 + \frac{{{{c}_{{12}}}}}{{{{c}_{{11}}}}} - 2{{{\left( {\frac{{{{c}_{{12}}}}}{{{{c}_{{11}}}}}} \right)}}^{2}}} \right]~$Цветные линии на диаграммах имеют следующий смысл.
Линии Коши (красные линии) соответствуют соотношению Коши. Если все атомы расположены в узлах кристаллической решетки, а межатомные силы являются центральными силами и могут быть описаны гармоническим потенциалом, то для упругих констант должны выполнятся соотношения Коши ${{c}_{{iijk}}} = {{c}_{{ijik}}}$ ($i \ne j,~k$) [28]. Для кубических кристаллов соотношения Коши сводятся к одному соотношению:
На диаграмме ${{c}_{{44}}}{\text{/}}{{c}_{{11}}} - {{c}_{{12}}}{\text{/}}{{c}_{{11}}}$ соотношение (2.14) выполняется на исходящей из центра координат биссектрисе угла между осями ${{c}_{{44}}}{\text{/}}{{c}_{{11}}}$ и ${{c}_{{12}}}{\text{/}}{{c}_{{11}}}$. Согласно литературе, например [29], многие из кубических кристаллов следуют именно этому тренду.
На диаграмме $A - {{\nu }_{0}}$ зависимость (2.14) имеет нелинейный вид
Отсюда и далее будем называть кривую, описываемую уравнением (2.15), кривой или линией Коши. На диаграмме $A - {{\nu }_{0}}$ кривая Коши начинается в правом верхнем углу (A = 1.5; ${{\nu }_{0}}$ = 0.5) и асимптотически приближается к ν0 = 0 при $A \to - \infty $.
Следует отметить, что разность ${{p}_{c}} = {{c}_{{12}}} - {{c}_{{44}}}$ называют давлением Коши. Поэтому линия, соответствующая соотношению Коши, разделяет диаграммы на две части: верхнюю, где давление Коши положительно (${{c}_{{12}}} > {{c}_{{44}}}$), и нижнюю, где давление Коши отрицательно (${{c}_{{12}}} < {{c}_{{44}}}$).
Линии Китинга (синие линии) теоретически предсказаны Китингом [30, 31] для алмазоподобных кристаллов с ковалентными связями методом наложения необходимых условий инвариантности на энергию деформации кристалла. В величинах упругих жесткостей cij оно имеет вид:
(2.16)
$2{{c}_{{44}}}\left( {{{c}_{{11}}} + {{c}_{{12}}}} \right) = \left( {{{c}_{{11}}} - {{c}_{{12}}}} \right)\left( {{{c}_{{11}}} + 3{{c}_{{12}}}} \right)$Поэтому как на диаграмме ${{c}_{{44}}}{\text{/}}{{c}_{{11}}} - {{c}_{{12}}}{\text{/}}{{c}_{{11}}}$ так и на диаграмме $A - {{\nu }_{0}}$ зависимость Китинга имеет нелинейный вид.
Линии Ледбеттера (зеленые линии) имеют значение подобное кривым Китинга, но эта зависимость получена эмпирически Ледбеттером и соавторами [29] путем линейной аппроксимации экспериментальных точек на диаграмме ${{c}_{{44}}}{\text{/}}{{c}_{{11}}} - {{c}_{{12}}}{\text{/}}{{c}_{{11}}}$, соответствующих одноатомным и двухатомным ковалентным кубическим кристаллам. В величинах упругих жесткостей cij уравнение этой линии имеет вид:
В координатах $A$ – ${{\nu }_{0}}$ зависимость Ледбеттера нелинейна:
Поэтому на диаграмме $A - {{\nu }_{0}}$ зеленая линия искривлена. Краткий вывод формул (2.13), (2.15), (2.17), (2.19) представлен в пункте 4.
Примечательно, что прямолинейная граница, разделяющая 1-ую и 2-ую характеристические области коэффициента Пуассона, и описываемая уравнением (2.12), расположена между кривыми Китинга и Ледбеттера, см. рис. 1,b. С уменьшением ν0 кривая Китинга плавно примыкает к этой границе в начале координат (A = 0; ${{\nu }_{0}}$ = 0), т.е. для обеих линий производная $d{{\nu }_{0}}{\text{/}}dA\left( {A = 0} \right) = 1{\text{/}}2$, а кривая Ледбеттера плавно приближается к данной границе при увеличении ν0 и касается ее в точке (A = 2/3; ${{\nu }_{0}}$ = 1/3) с идентичным же наклоном $d{{\nu }_{0}}{\text{/}}dA\left( {A = 2{\text{/}}3} \right) = 1{\text{/}}2$.
Достоинством широко известной диаграммы ${{c}_{{44}}}{\text{/}}{{c}_{{11}}} - {{c}_{{12}}}{\text{/}}{{c}_{{11}}}$ является ее простота. В то же время эта диаграмма неудобна для анализа величин коэффициента упругой анизотропии A и коэффициента Пуассона ν0. Поскольку именно эти характеристики упругости рассматриваются в настоящей работе, ниже будет использоваться диаграмма $A - {{\nu }_{0}}$, где переменными координатных осей являются A и ν0. Преимуществом последней диаграммы является также линейность границ, разделяющих характеристические области ауксетичности, что упрощает анализ и идентификацию типа ауксетиков.
3. Анализ зависимости упругих свойств от типа структуры и межатомной связи. На рис. 2 представлена диаграмма $A - {{\nu }_{0}}$ для кубических металлов с А1 структурой меди (ГЦК) и А2 структурой вольфрама (ОЦК). Каждая точка на диаграмме имеет координаты $\left( {A,{{\nu }_{0}}} \right)$, определяемые упругими свойствами металла, название которого указано рядом. Красные символы ▼, ◼, ◆, ⬢ соответствуют ГЦК металлам, синие символы ▲, ⚫ – ОЦК металлам. Разные обозначения точек соответствуют разным типам металлов: щелочным (Li, K, Na, Cs), редкоземельным (Ca, Sr), переходным (V, Cr, Fe, Ni, Cu, Nb, Mo, Rh, Pd, Ag, Ta, W, Ir, Pt, Au), постпереходным (Al, Pb), актиноидам (Ce, Yb) и лантаноидам (Th). Как видно из рис. 2 все ГЦК металлы положительно анизотропны (A > 0), тогда так часть ОЦК металлов, V, Cr, Nb и Mo, показывает отрицательную анизотропию (A < 0). ОЦК металл W практически изотропен ($A \approx 0$). Точки большинства металлов сконцентрированы в верхней правой части диаграммы, при этом их плотность особенно высока вблизи угла диаграммы (A = 1.5; ${{\nu }_{0}}$ = 0.5) в окрестности треугольной области, ограниченной кривой Коши и прямой ${{\nu }_{0}} = A{\text{/}}2$. Эти металлы являются частичными ауксетиками, так как их точки $\left( {A,{{\nu }_{0}}} \right)$ лежат правее граничной прямой ${{\nu }_{0}} = A{\text{/}}2$, т.е. в зоне ауксетичности 2. Наиболее приближены к углу диаграммы точки щелочных ОЦК металлов Li, K, Na и щелочноземельный ГЦК металл Sr. Эти металлы имеют наименьшее значение коэффициента Пуассона среди кубических кристаллов, представленных на рис. 2: ${{\nu }_{{{\text{min}}}}} = - 0.54$ у Li, ${{\nu }_{{{\text{min}}}}} = - 0.44$ у Na, ${{\nu }_{{{\text{min}}}}}$ = –0.42 у K, ${{\nu }_{{{\text{min}}}}} = - 0.26$ у Sr [15, 18]. Их коэффициент анизотропии A и коэффициент ${{\nu }_{0}}$ близки к максимально возможным значениям и соответственно находятся в интервалах 1.22–1.29 и 0.432–0.457. Несколько далее от угла диаграммы отстоят щелочноземельный ГЦК металл Ca и щелочной ОЦК металл Cs, которые имеют большие по абсолютной величине отрицательные минимальные значения коэффициента Пуассона (${{\nu }_{{{\text{min}}}}} = - 0.27$ у Ca и ${{\nu }_{{{\text{min}}}}} = - 0.36$ у Cs). В пределах вышеуказанной треугольной области располагаются такие переходные металлы как Cu, Ag, Au, Pd, Ni (все ГЦК) и Fe (ОЦК), и постпереходной ГЦК-металл Pb. У переходных ГЦК металлов минимальное значение коэффициента Пуассона не превосходит –0.13: ${{\nu }_{{{\text{min}}}}} = - 0.13$ у Cu, ${{\nu }_{{{\text{min}}}}} = - 0.13$ у Fe, ${{\nu }_{{{\text{min}}}}} = - 0.09$ у Ag, ${{\nu }_{{{\text{min}}}}} = - 0.07$ у Ni, ${{\nu }_{{{\text{min}}}}} = - 0.04$ у Pd и ${{\nu }_{{{\text{min}}}}} = - 0.03$ у Au. Постпереходный ОЦК-металл Pb имеет ${{\nu }_{{{\text{min}}}}} = - 0.20$. Актиноиды Ce (${{\nu }_{{{\text{min}}}}}$ = –0.17) и Yb (${{\nu }_{{{\text{min}}}}} = - 0.34$) и лантаноид Th (${{\nu }_{{{\text{min}}}}} = - 0.22$) (все ГЦК) находятся несколько ниже. Pt, Rh и Ir (ГЦК переходные металлы платиновой группы) сконцентрированы у пересечения кривых Коши и Китинга и являются неауксетиками.
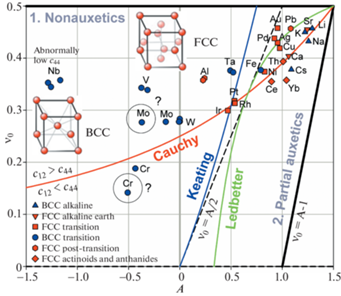
Характерно, что металлы из одной подгруппы периодической системы элементов, как правило, показывают подобные упругие свойства, а их точки подобным образом расположены на диаграмме $A - {{\nu }_{0}}$, например, см. конфигурации Li, K, Na, Cs (AI) и Cu, Ag, Au (BI) с одним валентным электроном, Ca, Sr (AII) с двумя валентными электронами, V, Nb, Ta (BV) и Cr, Mo, W (BVI) соответственно с пятью и шестью валентными электронами. Такое подобие упругих свойств закономерно, т.к. прочность металлической связи повышается с увеличением числа валентных электронов, участвующих в ее образовании.
Следует отметить, что после построения диаграммы на рис. 2 по данным справочника Ландольт–Берштайн [32] было обнаружено, что точки таких металлов как Ta, Al и Mo, и особенно V и Nb лежат существенно выше кривой Коши (область положительных “давлений Коши”, ${{c}_{{12}}} > {{c}_{{44}}}$), а Cr ниже, где ${{c}_{{12}}} < {{c}_{{44}}}$. Сравнение с данными из других литературных источников [33–37], см. множественные точки этих металлов, подтвердило достоверность данных для Ta, Al, V и Nb, но в то же время показало различие для Mo и Cr. Поэтому вызывающие сомнения точки Mo и Cr обозначены знаком “?”. Причиной смещения Ta, Al, V и Nb в область положительных “давлений Коши” является низкое значение их сдвиговой константы c44 по сравнению с константой ${{c}_{{12}}}$. Для этих металлов отношения ${{c}_{{44}}}{\text{/}}{{c}_{{12}}}$ соответственно равны: ≈0.53, ≈0.48, ≈0.39 и ≈0.23.
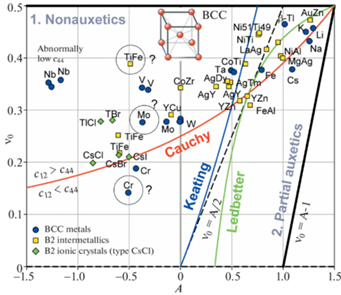
На рис. 3 сравниваются A и ${{\nu }_{0}}$ кристаллов, в которых атомы имеют ОЦК пространственное расположение в общей кристаллической решетке, но имеют разный тип межатомной связи: ОЦК-металлы – металлическая, В2 интерметаллиды – преимущественно металлическая, частично ионно-ковалентная и В2 ионные кристаллы – преимущественно ионная, частично ковалентная. Видно, что все точки, независимо от типа межатомной связи в кристаллах, имеют тенденцию располагаться вблизи кривой Коши, однако имеет место также общая закономерность смещения их вверх, где ${{c}_{{12}}} > {{c}_{{44}}}$. Это особенно явно выражено для металлов V и Nb, как отмечалось выше, а также для интерметаллида CoZr. Сильное смещение вверх одной из точек интерметаллида TiFe, помеченной знаком “?”, очевидно ошибочно, так как это не согласуется с двумя другими точками TiFe, расположенными вблизи кривой Коши, построенными по данным из двух независимых источников [32, 42]. Характерно, что В2 интерметаллиды, также как и ОЦК-металлы, могут быть положительно или отрицательно анизотропны. Большинство положительно анизотропны (A > 0), но некоторые, YCu и TiFe, отрицательно анизотропны (A < 0), а интерметаллид CoZr – изотропен ($A \approx 0$). Все представленные на рис. 3 В2 ионные кристаллы отрицательно анизотропны и являются неауксетиками, однако из этого видимо не следует делать вывод о том, что это свойство является общим для ионных кристаллов с В2 структурой, так как число представленных точек мало. Следует отметить, что число известных В2 ионных кристаллов (структура типа CsCl) весьма ограничено, например, CsCl, CsBr, TlCl, TlBr (представлены на рис. 3) и под высоким давлением RbCl, RbBr и RbI. Кристаллы со структурой типа CsCl являются соединениями одновалентных атомов металла и галогена. При этом ОЦК-расположение атомов возможно если атом металла достаточно большой, как, например, атом Cs. В противном случае отрицательные ионы галогенных атомов сойдутся на расстояние, при котором начнется их отталкивание.
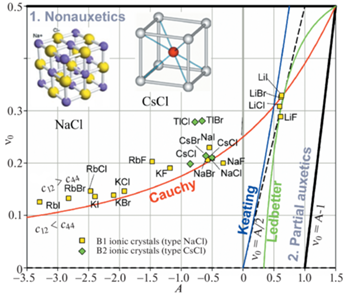
На рис. 4 сравниваются A и ${{\nu }_{0}}$ ионных кристаллов со структурой В1 и В2. В обоих случаях межатомная связь ионная, но разное пространственное расположение атомов. В случае В1 (структурный тип NaCl) атомы расположены в общей примитивной кристаллической решетке, в случае В2 (структурный тип CsCl) – в общей ОЦК-решетке. Видно, что точки, соответствующие большинству кристаллов, лежат вблизи кривой Коши, хотя и несколько смещены вверх, где ${{c}_{{12}}} > {{c}_{{44}}}$. Из общей тенденции несколько выпадают точки TlCl и TlBr, однако в отличие от остальных представленных на рис. 4 ионных соединений, в данных соединениях положительный ион Tl+ является не щелочным металлом, а полуметаллом. Также видно, что группа LiAM (AM = F, Cl, Br или I) имеет тенденцию быть выстроенной вдоль участка локального совпадения кривой Ледберга и прямой ${{\nu }_{0}} = A{\text{/}}2$, где должны располагаться точки ковалентных кристаллов. Причина этого, видимо, заключается в том, что у соединений LiX имеется небольшая доля ковалентной связи. Отметим, что в группе LiAM (AM = F, Cl, Br или I) ауксетические свойства проявляет только LiF (${{\nu }_{{{\text{min}}}}} = - 0.02$), т.к. фтор F располагается во 2-м периоде таблицы Д.И. Менделеева, тогда как Cl, Br и I соответственно в 3-м, 4-м и 5-м.
Рис. 4.
Сравнение A и ν0 кубических В1 и В2 ионных кристаллов, соответственно структурные типы NaCl и CsCl. Диаграмма построена с использованием упругих констант из [32].
На рис. 5 представлена диаграмма $A - {{\nu }_{0}}$ отдельно для кубических кристаллов со структурой типа NaCl (В1). Такую структуру образуют бинарные соединения AIBVII т.е. галогениды (фториды, хлориды, бромиды и иодиды) таких щелочных элементов как литий (LiF, LiCl, LiBr, LiI), натрий (NaF, NaCl, NaBr, NaI), калий (KF, KCl, KBr, KI) и рубидий (RbF, RbCl, RbBr, RbI), а также хлорид и бромид серебра (AgCl, AgBr). Кроме того, структуру типа NaCl имеют некоторые окислы металлов, например, MgO и CaO (бинарные соединения типа AIIBVI). Для каждой точки на рис. 5 приведена доля ионной связи fi, характеризующая степень поляризации межатомной связи. Данные значения fi были рассчитаны Филлипсом [45] с использованием спектроскопической теории межатомной связи. Проведенное Филлипсом сравнение с результатами предыдущих термохимических расчетов fi, проведенных Полингом [49], и расчетов Кулсона с соавторами [50], основанных на концепции валентных связей, показало в целом удовлетворительное совпадение. На рис. 5 видна тенденция: при смещении вверх по кривой Коши доля ионной связи fi уменьшается. Например, RbI с fi = 0.951 находится в нижней части кривой Коши, а LiF с fi = 0.89 в верхней.
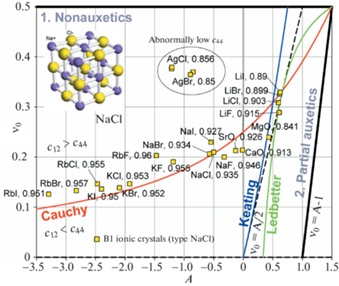
Точки ($A,~{{\nu }_{0}}$), соответствующие соединениям с fi > 0.9 довольно хорошо ложатся на кривую Коши, тогда как для соединений с fi ≈ 0.85, MgO, AgCl и AgBr имеется отклонение от этой кривой. В особенности оно велико для хлорида и бромида серебра, коэффициент Пуассона которых ${{\nu }_{0}}$ ≈ 0.37, значительно выше, чем у других ионных кристаллов, а также низкая величина сдвиговой константа ${{c}_{{44}}}$. Достоверность приведенных на рис. 5 значений для AgCl и AgBr подтверждается сравнением данных из различных источников [32, 46, 47], см. двойные точки.
Из рис. 5 видно, что наибольшая отрицательная анизотропия, –3.3 < A < –1.5, и, соответственно, наименьший коэффициент Пуассона, 0.13 < ${{\nu }_{0}}$ < 0.2, наблюдаются для галогенидов рубидия. Для этих соединений также типична высокая поляризация ионной связи, fi ≈ 0.95. Для галогенидов лития наблюдается положительная анизотропия, $A$ ≈ +0.6, и, соответственно, наибольший коэффициент Пуассона, ${{\nu }_{0}}$ ≈ 0.29–0.33. Для этих соединений также типична более низкая доля ионной связи, fi ≈ 0.9. Точки для галогенидов калия и натрия занимают промежуточные значения как по величинам A и ${{\nu }_{0}}$, так и по величине fi. В целом имеется следующая общая тенденция: при смещении щелочно-металлического компонента вниз в подгруппе АI периодической таблицы элементов Li → Na → K → Rb, т.е. с увеличением номера периода, точки ($A,~{{\nu }_{0}}$) смещаются вниз по кривой Коши. Зависимость от типа элемента-галогена не столь явная, но в первом приближении для них наблюдается обратная тенденция, с увеличением номера периода F → Cl → Br → I точки ($A,~{{\nu }_{0}}$) смещаются вверх по кривой Коши.
На рис. 6 представлена диаграмма $A - {{\nu }_{0}}$ для кристаллов со структурой алмаза А4, это элементы AIV группы периодической системы, C, Si, Ge и α-Sn, и кристаллов с алмазоподобной структурой цинковой обманки В3, в основном – бинарные соединения типа ANB8–N. Из представленных кристаллов на рис. 6 это:
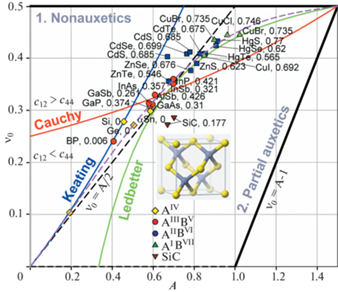
– AIIIBV – борид фосфора BP, фосфиды, арсениды и антимониды алюминия, галия и индия, соответственно AlSb, GaP, GaAs, GaSb, и InP, InAs, InSb. Представленные материалы являются неауксетиками.
– AIIBVI – сульфиды, селениды и теллуриды цинка, кадмия и ртути, соответственно ZnS, ZnSe, ZnTe; CdS, CdSe, CdTe и HgS, HgSe, HgTe. Частичными ауксетиками являются ZnSe, ZnTe, CdTe и HgS, HgSe, HgTe [15, 18].
– AIBVII – хлориды, бориды и иодиды меди, соответственно CuCl, CuBr и CuI. Данные кристаллы являются частичными ауксетиками.
Также структуру цинковой обманки имеют кристаллы карбида кремния SiC. Кристалл SiC является частичным ауксетиком (${{\nu }_{{{\text{min}}}}} = - 0.10$).
На рис. 6 видно, что все алмазоподобные кристаллы положительно анизотропны (A > 0) и для них наблюдается зависимость: с повышением доли ионной связи fi упругая анизотропия, характеризуемая коэффициентом $A$, возрастает, и соответственно повышается значение ν0. Так для алмаза с fi = 0, расположенного вблизи нижнего левого угла диаграммы, $A \approx $ 0.19 и ${{\nu }_{0}} \approx 0.10$, для арсенида галия GaAs с ${{f}_{i}} = $ 0.31, расположенного центральной части диаграммы, $A \approx $ 0.60 и ${{\nu }_{0}} \approx 0.31$, а для бромида меди CuBr с fi = 0.735, расположенного в вблизи верхнего правого угла диаграммы, $A \approx $ 1.02 и ${{\nu }_{0}} \approx 0.44$.
Из рис. 6 видно, что точки, соответствующие полностью ковалентным алмазу C и кремнию Si, а также почти полностью ковалентному бориду фосфора BP (${{f}_{i}} = $ 0.006) хорошо ложатся на теоретическую кривую Китинга. Однако для двух других полностью ковалентных кристаллов, германия Ge и олова α-Sn имеется заметное отклонение от этой зависимости в сторону бóльших значений коэффициента упругой анизотропии A. По мере увеличения fi это отклонение ΔA возрастает. Так для AIIIBV соединений, группы GaP, GaAs, GaSb, и InP, InAs, InSb, с fi ≈ 0.3–0.4 отклонение ΔA составляет около 0.1–0.12, тогда как для AIBVII соединений, CuCl и CuBr, с fi ≈ 0.75 отклонение ΔA достигает ≈0.3. Соединения AIIBVI, ZnS, ZnSe, ZnTe; CdS, CdSe, CdTe и HgS, HgSe, HgTe, с fi ≈ 0.55–0.7 занимают промежуточное положение, для них ΔA ≈ 0.2.
Примечательным является тот факт, что точки большинства А4 и В3 кристаллов расположены вблизи прямой линии $~{{\nu }_{0}} = A{\text{/}}2$ являющейся границей, разделяющей характеристические области “1. Неауксетики” / “2. Частичные ауксетики”. Однако с повышением fi наблюдается отклонение и от этой граничной линии, т.е. кристаллы становятся частичными ауксетиками, как, например, HgTe (${{\nu }_{{{\text{min}}}}} = - 0.03$), HgSe (${{\nu }_{{{\text{min}}}}}$ = –0.05), HgS (${{\nu }_{{{\text{min}}}}} = - 0.04$) и CuI (${{\nu }_{{{\text{min}}}}} = - 0.03$), CuCl (${{\nu }_{{{\text{min}}}}} = - 0.07$) и CuBr (${{\nu }_{{{\text{min}}}}}$ = –0.03). Эмпирическая зависимость Ледбеттера удовлетворительно описывает отклонение точек от прямой ${{\nu }_{0}} = A{\text{/}}2$ в верхней правой части диаграммы, но дает большую ошибку в нижней левой ее части для таких полностью ковалентных кристаллов как алмаз C, борид фосфора BP и кремний Si. Поэтому для описания положения точек А4 и В3 кристаллов в настоящей работе предлагается зависимость:
Данная зависимость была получена при следующих ограничениях: исход из начала координат ($A = 0$, ν0 = 0), также как теоретическая кривая Китинга и граничная прямая ${{\nu }_{0}} = A{\text{/}}2$, с таким же наклоном – $d{{\nu }_{0}}{\text{/}}dA\left( {A = 0} \right) = 0.5$. Окончание – в углу диаграммы ($A = 1.5$, ν0= 0.5), также как и у кривой Ледбеттера. Последнее ограничение накладывается на подгоночные коэффициенты при A2, A3 и A4, поэтому только два из них являются независимыми и определяют форму кривой ${{\nu }_{0}} = f\left( A \right)$. На рис. 6 зависимость (3.1) показана штриховой фиолетовой линией. Видно, что данная кривая правильно отражает характер зависимости ${{\nu }_{0}} = f\left( A \right)$ и удовлетворительно описывает положение большинства точек.
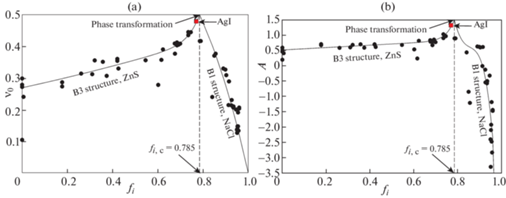
На рис. 7 представлены графики зависимостей коэффициента упругой анизотропии A и коэффициента Пуассона ${{\nu }_{0}}$ кристаллов со структурой В1 и В3 от доли ионной связи fi. На графиках видно, что с повышением доли ионной связи fi коэффициенты $A$ и ${{\nu }_{0}}$ сначала повышаются достигая максимальных значений Amax и ${{\nu }_{{0,{\text{max}}}}}$ при критическом значении ${{f}_{i}} = {{f}_{{i,c}}}$, а затем резко понижаются. Исходя из графика на рис. 5 логично предположить, что A и ${{\nu }_{0}}$ стремятся соответственно к $ - \infty $ и 0 при ${{f}_{i}} \to 1$. На основании наблюдаемых на рис. 6 экспериментальных трендов и формы кривых Коши и Ледбеттера можно также предположить, что ${{A}_{{{\text{max}}}}}$ = 1.5 и ${{\nu }_{{0,{\text{max}}}}}$ = 0.5, т.е. их значения совпадают с координатами предельной точки $\left( {A,{{\nu }_{0}}} \right)$ в верхнем правом углу диаграммы $A - {{\nu }_{0}}$. Наиболее близка к этим значениям красная точка В3 кристалла йодистого серебра AgI с $A \approx 1.3$ и ${{\nu }_{0}} \approx 0.48$. Данные величины получены используя константы cij, рассчитанные для В3 структуры AgI модифицированным методом Мартина в [44]. Показательно, что все точки слева от максимума соответствуют В3-кристаллам (структура ZnS), тогда как все точки справа – В1-кристаллам (структура NaCl). Из этого следует, что при достижении критической доли ионной связи ${{f}_{i}} = {{f}_{{i,c}}}$ происходит изменение кристаллической структуры В1 → В3. Согласно Филлипсу [45] это ковалентно-ионное превращение происходит при ${{f}_{{i,c}}} = 0.785$, что соответствует положению максимумов на графиках, представленных на рис. 7. Характерно, что кристаллы с fi близким к 0.785, например, AgI, MgS и MgSe структурно нестабильны, см. [46, 47].
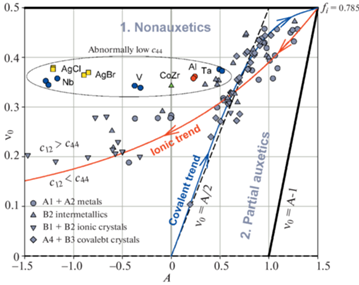
На рис. 8 представлена диаграмма $A~ - {{\nu }_{0}}$, обобщающая приведенные выше результаты для разных типов одноатомных и двухатомных кубических кристаллов. На диаграмме показаны два тренда – ковалентный (синяя линия, аппроксимация (3.1)) и ионный (красная линия, зависимость Коши (2.14, 2.15)). Исключая из рассмотрения особые точки (выделены цветом), можно сделать вывод о том, что все остальные точки (серые) в первом приближении следуют этим трендам. При этом для точек, прилегающих к линии Коши, имеется систематическое смещение вверх от этой линии, т.е. для них выполнятся неравенство ${{c}_{{12}}} > {{c}_{{44}}}$. Для некоторых кристаллов, например, Ta, Al, CoZr, V, AgBr, AgCl, Nb (см. цветные точки, заключенных в овал) это смещение особенно велико потому, что величина их сдвиговой константы c44 аномально низка. Замечательным свойством диаграммы $A~ - {{\nu }_{0}}$ является то, что в отличие от диаграммы Блекмана (рис. 1,а) на ней оба тренда образуют единую непрерывную линию ${{\nu }_{0}} = f\left( A \right)$, которая исходит из начала координат $\left( {A = 0,~\,\,{{\nu }_{0}} = 0} \right)$, далее идет вверх вправо и приходит в экстремальную угловую точку диаграммы $\left( {A = 1.5,\,\,{{\nu }_{0}} = 0.5} \right)$, а затем изгибается и идет вниз влево стремясь в пределе к $\left( {A = - \infty ,\,\,{{\nu }_{0}} = 0} \right)$. Причиной этого различия в представлении кривых является то, что вся верхняя горизонтальная граница диаграммы Блекмана (${{c}_{{12}}}{\text{/}}{{c}_{{11}}} = 1$), где заканчиваются линии Коши и Ледбеттера, сводится на диаграмме $A~ - {{\nu }_{0}}$ в одну точку $\left( {A = 1.5,~\,\,{{\nu }_{0}} = 0.5} \right)$. Положение точек $\left( {A,~{{\nu }_{0}}} \right)$ ионно-ковалентных кристаллов на единой кривой ${{\nu }_{0}} = f\left( A \right)$ определяется величиной доли ионной связи fi. При увеличении fi от нуля до критического значения 0.785 происходит смещение точек $\left( {A,~{{\nu }_{0}}} \right)$ вдоль ковалентного тренда от начала координат к угловой точке $\left( {A = 1.5,~{{\nu }_{0}} = 0.5} \right)$. При дальнейшем увеличении fi точки переходят на ионный тренд и смещаются в направлении предела $\left( {A = - \infty ,~{{\nu }_{0}} = 0} \right)$. Следует отметить, что перегиб единой кривой ${{\nu }_{0}} = f\left( A \right)$ в точке $\left( {A = 1.5,~\,\,{{\nu }_{0}} = 0.5} \right)$ согласуется с выводами работы [44], согласно которой ковалентно-ионный переход (изменение структуры) происходит вследствие нестабильности кристаллической решетки при приближении эффективного модуля сдвига ${{c}_{s}} = \frac{1}{2}\left( {{{c}_{{11}}} - {{c}_{{12}}}} \right)$ к нулю, что соответствует стремлению коэффициента Пуассона к пределу ${{\nu }_{0}} \to 0.5$.
Рис. 8.
Обобщенная диаграмма $A~ - {{\nu }_{0}}$ для разных типов одноатомных и двухатомных кубических кристаллов.
4. Вывод формул (2.13), (2.15), (2.17) и (2.19). Уравнение негоризонтальной границы характеристических областей коэффициента Пуассона
Подставляя A и ν0 соответственно из (2.4) и (2.8), и используя (2.1), (2.2) и (2.3) с заменой sij → cij, получаем
(4.2)
$\frac{{{{c}_{{44}}}}}{{{{c}_{{11}}}}} = \frac{1}{2}\left[ {1 + \frac{{{{c}_{{12}}}}}{{{{c}_{{11}}}}} - 2{{{\left( {\frac{{{{c}_{{12}}}}}{{{{c}_{{11}}}}}} \right)}}^{2}}} \right]{\text{\;}}$Соотношение Коши
Поставляя c12 и c44 соответственно из (2.2) и (2.3), и учитывая (2.4) и (2.8) получаем квадратное уравнение относительно ${{{{\nu }}}_{0}}$
Его физически корректное решение
Соотношение Китинга
(4.6)
$2{{c}_{{44}}}\left( {{{c}_{{11}}} + {{c}_{{12}}}} \right) = \left( {{{c}_{{11}}} - {{c}_{{12}}}} \right)\left( {{{c}_{{11}}} + 3{{c}_{{12}}}} \right)$Поставляя c11, c12 и c44 соответственно из (2.1), (2.2) и (2.3), а также учитывая (2.4) и (2.8) получаем квадратное уравнение
Его физически корректное решение
Соотношение Ледбеттера
Проводя аналогичные подстановки как в предыдущем случае, получаем квадратное уравнение
Его физически корректное решение
5. Заключение. С использованием диаграммы $A~ - {{\nu }_{0}}$ проанализирована зависимость упругих свойств одноатомных и двухатомных кубических кристаллов от типов их кристаллической структуры и межатомной связи. Показано, что упругие свойства этих кристаллов следуют одному из двух трендов – ковалентному или ионному, которые на диаграмме $A~ - {{\nu }_{0}}$ образуют единую непрерывную линию ${{\nu }_{0}}\, = \,f(A)$, исходящую из начала координат $(A = 0,~{{\nu }_{0}} = 0)$, приходящую в угловую точку диаграммы $(A = 1.5,~\,\,{{\nu }_{0}} = 0.5)$ и далее изгибаясь стремящуюся к $(A = - \infty ,~\,\,{{\nu }_{0}} = 0)$. Смещение точек $(A,~{{\nu }_{0}})$ ионно-ковалентных кристаллов вдоль этой линии определяется величиной доли ионной связи fi. При $0 \leqslant {{f}_{i}} < 0.785$ точки $(A,~{{\nu }_{0}})$ располагаются на ковалентном тренде, а при ${{f}_{i}} > 0.785$ – на ионном.
Проведенный анализ показал, что ауксетики выявляются среди одноатомных кристаллов со структурой А1 (ГЦК), относящимся к щелочноземельным (Ca, Sr), переходным (Ni, Cu, Ag, Au) и постпереходным (Pb) металлам, актиноидам (Ce, Yb) и лантаноидам (Th), и одноатомных кристаллов со структурой А2 (ОЦК), относящимся к щелочным металлам (Li, K, Na, Cs). Наибольшие отрицательные значения коэффициента Пуассона наблюдаются у щелочных металлов (${{\nu }_{{{\text{min}}}}} = - 0.54$ у Li, ${{\nu }_{{{\text{min}}}}} = - 0.44$ у Na, ${{\nu }_{{{\text{min}}}}} = - 0.42$ у K, ${{\nu }_{{{\text{min}}}}} = - 0.26$ у Sr). Большие отрицательные значения коэффициента Пуассона выявляются у щелочноземельных металлов (${{\nu }_{{{\text{min}}}}} = - 0.27$ у Ca и ${{\nu }_{{{\text{min}}}}}$ = = –0.36 у Cs), постпереходного металла Pb (${{\nu }_{{{\text{min}}}}} = - 0.20$), актиноидов Ce (${{\nu }_{{{\text{min}}}}}$ = –0.17), Yb (${{\nu }_{{{\text{min}}}}} = - 0.34$) и лантаноида Th (${{\nu }_{{{\text{min}}}}} = - 0.22$).
Среди проанализированных в этой работе бинарных двухатомных соединений наибольшее количество ауксетиков выявляется среди алмазоподобных кристаллов со структурой цинковой обманки B3, имеющих стехиометрию типа ANB8-N (AIIBVI: ZnSe, ZnTe, CdTe и HgS, HgSe, HgTe; AIIBVI: CuCl, CuBr и CuI). Кристаллы соединений типа AIIIBV, являются неауксетиками. Среди кубических В1 и В2 ионных кристаллов, структурных типов NaCl и CsCl, только один LiF, по-видимому, является ауксетиком с небольшим по абсолютной величине ${{\nu }_{{{\text{min}}}}} = - 0.02$.
Работа выполнена при поддержке гранта Российского научного фонда (проект № 22-29-01200).
Список литературы
Най Дж. Физические свойства кристаллов и их описание при помощи тензоров и матриц. М.: Мир, 1967. 385 с.
Cazzani A., Rovati M. Extrema of Young’s modulus for cubic and transversely isotropic solids // Int. J. Solids Struct. 2003. V. 40. № 7. P. 1713–1744.https://doi.org/10.1016/S0020-7683(02)00668-6
Гольдштейн Р.В., Городцов В.А., Лисовенко Д.С. Модуль Юнга кубических ауксетиков // Письма о материалах. 2011. Т. 1. Вып. 3. С. 127–132. https://doi.org/10.22226/2410-3535-2011-3-127-132f
Гольдштейн Р.В., Городцов В.А., Лисовенко Д.С. Модуль сдвига кубических кристаллов. Письма о материалах. 2012. Т. 2. Вып. 1. С. 21–24. https://doi.org/10.22226/2410-3535-2012-1-21-24
Hayes M., Shuvalov A. On the extreme values of Young’s modulus, the shear modulus, and Poisson’s ratio for cubic materials // J. Appl. Mech. 1998. V. 65. № 3. P. 786–787. https://doi.org/10.1115/1.2789130
Ting T.C.T., Chen T. Poisson’s ratio for anisotropic elastic materials can have no bounds // Quart. J. Mech. Appl. Math. 2005. V. 58. № 1. P. 73–82. https://doi.org/10.1093/qjmamj/hbh021
Milstein F., Huang K. Existence of a negative Poisson ratio in fcc crystals // Phys. Rev. B. 1979. V. 19. № 4. P. 2030–2033. https://doi.org/10.1103/PhysRevB.19.2030
Светлов И.Л., Епишин А.И., Кривко А.И., Самойлов А.И., Одинцев И.Н., Андреев А.П. Анизотропия коэффициента Пуассона монокристаллов никелевого сплава // ДАН СССР. 1988. Т. 302. № 2. С. 1372–1375.
Ballato A. Poisson’s ratio for tetragonal, hexagonal, and cubic crystals // IEEE Trans. Ultrason. Ferroelectrics Frequency Contr. 1996. V. 43. № 1. P. 56–62. https://doi.org/10.1109/58.484463
Baughman R.H., Shacklette J.M., Zakhidov A.A., Stafström S. Negative Poisson’s ratios as a common feature of cubic metals // Nature. 1998. V. 392. № 6674. P. 362–365. https://doi.org/10.1038/32842
Ting T.C.T., Barnett D.M. Negative Poisson’s ratios in anisotropic linear elastic media // J. Appl. Mech. 2005. V. 72. № 6. P. 929–931. https://doi.org/10.1115/1.2042483
Norris A.N. Poisson’s ratio in cubic materials // Proc. Roy. Soc. A. 2006. V. 462. № 2075. P. 3385–3405. https://doi.org/10.1098/rspa.2006.1726
Lethbridge Z.A.D., Walton R.I., Marmier A.S.H. et al. Elastic anisotropy and extreme Poisson’s ratios in single crystals // Acta Mater. 2010. V. 58. № 19. P. 6444–6451. https://doi.org/10.1016/j.actamat.2010.08.006
Branka A.C., Heyes D.M., Wojciechowski K.W. Auxeticity of cubic materials // Phys. Status Solidi B. 2009. V. 246. № 9. P. 2063–2071. https://doi.org/10.1002/pssb.200982037
Goldstein R.V., Gorodtsov V.A., Lisovenko D.S. Classification of cubic auxetics // Phys. Status Solidi B. 2013. V. 250. № 10. P. 2038–2043. https://doi.org/10.1002/pssb.201384233
Епишин А.И., Лисовенко Д.С. Экстремальные значения коэффициента Пуассона кубических кристаллов // ЖТФ. 2016. Т. 16. № 10. С. 1516–1524. https://doi.org/10.1134/S106378421610012
Гольдштейн Р.В., Городцов В.А., Лисовенко Д.С. Ауксетическая механика кристаллических материалов // Изв. РАН. МТТ. 2010. № 4. С. 43–62.
Городцов В.А., Лисовенко Д.С. Ауксетики среди материалов с кубической анизотропией // Изв. РАН. МТТ. 2020. № 4. С. 7–24. https://doi.org/10.31857/S0572329920040054
Paszkiewicz T., Wolski S. Anisotropic properties of mechanical characteristics and auxeticity of cubic crystalline media // Phys. Status Solidi B. 2007. V. 244. № 3. P. 966–977. https://doi.org/10.1002/pssb.200572715
Paszkiewicz T., Wolski S. Elastic properties of cubic crystals: Every’s versus Blackman’s diagram // J. Phys. Conf. Ser. 2008. V. 104. P. 012038. https://doi.org/10.1088/1742-6596/104/1/012038
Гольдштейн Р.В., Городцов В.А., Лисовенко Д.С. Связь среднего коэффициента Пуассона с модулем Юнга для кубических кристаллов. Ауксетики в среднем // ДАН. 2012. Т. 443. № 6. С. 677–681.
Evans K., Nkansah M., Hutchinson I., Rogers S.C. Molecular network design // Nature. 1991. V. 353. № 6340. P. 124. https://doi.org/10.1038/353124a0
Lim T.C. Auxetic Materials and Structures. Singapore: Springer Singapore, 2015. 588 p. https://doi.org/10.1007/978-981-287-275-3
Kadic M., Milton G.W., van Hecke M., Wegener M. 3D metamaterials // Nature Rev. Phys. 2019. V. 1. P. 198–210. https://doi.org/10.1038/s42254-018-0018-y
Kelkar P.U., Kim H.S., Cho K.-H. et al. Cellular auxetic structures for mechanical metamaterials: A review // Sensors. 2020. V. 20. P. 3132. https://doi.org/10.3390/s20113132
Luo C., Han C.Z., Zhang X.Y. et al. Design, manufacturing and applications of auxetic tubular structures: A review // Thin-Walled Struct. 2021. V. 163. P. 107682. https://doi.org/10.1016/j.tws.2021.107682
Blackman M. On anomalous vibrational spectra // Proc. Roy. Soc. A. 1938. V. 164. P. 62–79. https://doi.org/10.1098/rspa.1938.0005
Haussühl S. Kristallphysik. Weinheim: Physik-Verlag, 1983. p.
Ledbetter H. Blackman diagrams and elastic-constant systematics / Handbook of Elastic properties of solids, liquids, and gases. Ed. by M. Levy, H. Bass, and R. Stern. San Diego: Academic Press, 2000. Vol. II. P. 57–64. https://doi.org/10.1016/B978-012445760-7/50029-0
Keating P.N. Effect of invariance requirements on the elastic strain energy of crystals with application to the diamond structure // Phys. Rev. 1966. V. 145. № 2. P. 637–645. https://doi.org/10.1103/PhysRev.145.637
Keating P.N. Theory of the third-order elastic constants of diamond-like crystals // Phys. Rev. 1966. V. 149. P. 674–678. https://doi.org/10.1103/PhysRev.149.674
Second and Higher Order Elastic Constants / Ed. by D. F. Nelson. Springer, 1992. https://doi.org/10.1007/b44185
Vallin J., Mongy M., Salama K., Beckman O. Elastic constants of aluminum // J. Appl. Phys. 1964. V. 35. № 6. P. 1825–1826. https://doi.org/10.1063/1.1713749
Carroll K.J. Elastic constants of niobium from 4.2° to 300°K // J. Appl. Phys. 1965. V. 36. P. 3689–3690. https://doi.org/10.1063/1.1703072
Greiner J.D., Carlson O.N., Smith J.F. Single-crystal elastic constants of vanadium and vanadium with oxygen additions // J. Appl. Phys. 1979. V. 50. P. 4394–4398. https://doi.org/10.1063/1.326428
Palmer S.B., Lee E.W. The elastic constants of chromium // Philos. Mag. 1971. V. 24. № 188. P. 311–318. https://doi.org/10.1080/14786437108227390
Zheng S., Wang S. First-principles design of refractory high entropy alloy VMoNbTaW // Entropy. 2018. V. 20. P. 965. https://doi.org/10.1080/14786437108227390
Sekkal A., Benzair A., Ouahrani T. et al. Mechanical properties and bonding feature of the YAg, CeAg, HoCu, LaAg, LaZn, and LaMg rare-earth intermetallic compounds: An ab initio study // Intermetallics. 2014. V. 45. P. 65–70. https://doi.org/10.1016/j.intermet.2013.10.007
Liu L., Wu X., Li W. et al. High temperature and pressure effects on the elastic properties of B2 intermetallics AgRE // Open Phys. 2015. V. 13. № 1. P. 142–150. https://doi.org/10.1515/phys-2015-0019
Lu W., Li C., Yi J., Li K. Stability and elastic properties of B2 CoX (X = Ti, Zr and Hf) intermetallic compounds as a function of pressure // Philos. Mag. 2018. V. 98. № 3. P. 203–218. https://doi.org/10.1080/14786435.2017.1400701
Chen Y., Yao Z.J., Zhang P.Z. et al. Elastic constants and properties of B2-type FeAl and Fe–Cr–Al alloys from first-principles calculations // AER. 2016. V. 85. P. 380–386. https://doi.org/10.2991/ame-16.2016.63
Muslov S.A., Lotkov A.I. Extremes of the elasticity characteristics of TiFe and TiNi single crystals // AIP Conf. Proc. 2018. V. 2051. P. 020207. https://doi.org/10.1063/1.5083450
Morris J.R., Ye Y.Y. Phase stability, elastic constants, and defect energetics in ductile ordered b2 compounds / Ductile Rare Earth Intermetallic Compounds. 2005. P. 133–134. https://corpora.tika.apache.org/base/docs/govdocs1/230/230790.pdf
Morris J.R., Ye Y.Y., Lee Y.-B. et al. Ab initio calculation of bulk and defect properties of ductile rare-earth intermetallic compounds // Acta Mater. 2004. V. 52. № 16. P. 4849–4857. https://doi.org/10.1016/j.actamat.2004.06.050
Phillips J.C. Ionicity of the chemical bonds in crystals // Rev. Modern Phys. 1970. V. 42. № 3. P. 317–356. https://doi.org/10.1103/RevModPhys.42.317
Hidshaw W., Lewis J.T., Briscoe C.V. Elastic constants of silver chloride from 4.2 to 300°K // Phys. Rev. 1967. V. 163. № 3. P. 876–881. https://doi.org/10.1103/PhysRev.163.876
Materials Data Silver Bromide (AgBr) – Crystran. https://www.crystran.co.uk/optical-materials/silver-bromide-agbr
Fjeldly T.A., Hanson R.C. Elastic and piezoelectric constants of silver-iodide: Study of a material at the covalent-ionic phase transition // Phys. Rev. B. 1974. V. 10. № 8. P. 3569–3577. https://doi.org/10.1103/PhysRevB.10.3569
Pauling L. The nature of the chemical bond and the structure of molecules and crystals. Ithaca, N.Y.: Cornell Univ. Press, 1940. 450 p.
Coulson C.A., Redei L.B., Stocker D. The electronic properties of tetrahedral intermetallic compounds I. Charge distribution // Proc. Roy. Soc. A. 1972. V. 270. P. 357–372. https://doi.org/10.1098/rspa.1962.0229
Souadkia M., Bennecer B., Kalarasse F. Elastic, vibrational and thermodynamic properties of α-Sn based group IV semiconductors and GeC under pressure // J. Phys. Chem. Solids. 2013. V. 74. P. 1615–1625. https://doi.org/10.1016/j.jpcs.2013.06.005
Adachi S. Properties of group-IV, III–V and II–VI semiconductors. John Wiley & Sons, 2005. 385 p. https://doi.org/10.1002/0470090340
Phillips J.C. Covalent-ionic and covalent-metallic transitions of tetrahedrally coordinated ANBN–8 crystals under pressure // Phys. Rev. Lett. 1971. V. 27. P. 1197–1200. https://doi.org/10.1103/PhysRevLett.27.1197
Vukić M.R., Veselinović D.S., Marković V.G. Crystalline forms of silver iodide II. Determination of phase transformations // J. Serb. Chem. Soc. 2007. V. 72. № 8–9. P. 857–868. https://doi.org/10.2298/JSC0709857V
Tairi L., Touam S., Boumaza A. et al. Phase stability and electronic behavior of MgS, MgSe and MgTe compounds // Phase Transitions. 2017. V. 90. № 10. P. 1–13. https://doi.org/10.1080/01411594.2017.1302085
Дополнительные материалы отсутствуют.
Инструменты
Известия РАН. Механика твердого тела