

Кинетика и катализ, 2020, T. 61, № 2, стр. 168-194
Каталитическое действие слабосвязанного окружения молекулярного кислорода в процессах фотоокисления
А. В. Бакланов a, b, *, Д. Паркер c
a ФГБУН Институт химической кинетики и горения им. В.В. Воеводского СО РАН
630090 Новосибирск, ул. Институтская, 3, Россия
b ФГБОУ ВПО Новосибирский Государственный Университет
630090 Новосибирск, ул. Пирогова, 2, Россия
c Неймегенский Университет Радбауд
6525 Неймеген, ул. Хейендаалсвег, 135, Нидерланды
* E-mail: baklanov@kinetics.nsc.ru
Поступила в редакцию 01.11.2019
После доработки 01.11.2019
Принята к публикации 05.11.2019
Аннотация
В настоящем обзоре в исторической последовательности рассматриваются спектральные и фотохимические проявления каталитического влияния слабосвязанного окружения на фотопроцессы с участием кислорода в газе, растворах и на поверхности. Проанализирован прогресс в экспериментальном изучении природы супрамолекулярной фотохимии слабосвязанных комплексов кислорода, связанный с использованием в качестве модельной системы Ван-дер-Ваальсовых комплексов X–O2, генерируемых в молекулярных пучках. Представлен обзор результатов изучения супрамолекулярной фотохимии в комплексах X–O2 с применением техники измерения карт скоростей фотофрагментов. Обсуждается природа драматического усиления УФ-поглощения слабосвязанными комплексами кислорода и механизм фотохимических процессов, к которым приводит усиленное поглощение. Обобщены имеющиеся экспериментальные результаты и сделанные на их основе выводы о природе полос поглощения, появляющихся в слабосвязанных комплексах X–O2, возбуждение в которых приводит к образованию синглетного кислорода и других интермедиатов фотоокисления.
СОДЕРЖАНИЕ
2. Спектральные и фотохимические проявления влияния слабосвязанного окружения на фотопроцессы с участием кислорода
2.1. Влияние слабосвязанного окружения на спектры поглощения кислорода
2.2. Влияние слабосвязанного окружения кислорода на процессы фотоокисления
3. Супрамолекулярная фотохимия Ван-дер-Ваальсовых комплексов X–O2
3.1. Ван-дер-Ваальсов комплекс X–O2 как модельная система. Первые эксперименты
3.2. Техника измерения карт скоростей фотофрагментов
3.3. Карты скоростей атомов кислорода O(3PJ)
3.4. Природа возбужденных состояний и механизмы супрамолекулярной фотохимии комплексов X–O2
1. ВВЕДЕНИЕ
Фотопроцессы с участием молекулярного кислорода являются ключевыми в разнообразных реакциях фотоокисления, протекающих в природе, в лабораторной и технологической практике. В силу высокой симметрии кислорода все оптические переходы в этой молекуле, которые могут осуществляться под действием излучения инфракрасного, видимого и ультрафиолетового диапазонов, запрещены по симметрии и/или спину, в результате чего значения сечения поглощения фотона очень малы. Например, поглощение молекулами кислорода излучения в континууме Герцберга (200–242 нм), которое сопровождается фотодиссоциацией на атомы, характеризуется сечениями поглощения в 106–107 раз меньшими, чем сечения разрешенных переходов. Ясно, что если есть факторы, которые могут ослаблять или снимать этот запрет, то они будут влиять на скорость и механизм фотоокисления. Оказывается, это происходит под действием молекулярного, в том числе химически инертного, окружения в газе или конденсированной среде. Возмущающее влияние взаимодействия молекулы кислорода с молекулами окружения при столкновениях в газовой фазе, в контактных комплексах в растворе, в твердой матрице или на поверхности приводит к драматическому усилению поглощения. Рост сечения поглощения на обсуждаемых переходах может достигать 6–7 порядков величины. Наряду с этим возникают новые полосы супрамолекулярного поглощения, где поглощающей частицей является слабосвязанный комплекс кислорода с молекулами окружения X–O2. При возбуждении в такой полосе образуется реакционноспособный синглетный кислород. Молекулы окружения при этом могут оставаться неизменными, выполняя роль фотокатализатора, либо могут также претерпевать химические изменения. Критически важным для достижения прогресса в понимании этих процессов в течение последние примерно 20 лет стало использование в качестве модельной системы Ван-дер-Ваальсовых комплексов кислорода X–O2, которые можно генерировать с помощью техники молекулярных пучков. В условиях молекулярного пучка стало возможным применение очень мощного метода, каким является техника измерения карт скоростей (velocity map imaging) фотофрагментов, использование которой позволило идентифицировать природу возбужденных электронных состояний комплексов, ответственных за супрамолекулярную фотохимию Ван-дер-Ваальсовых комплексов кислорода X–O2.
В обзоре в исторической последовательности рассмотрены спектральные и фотохимические проявления влияния слабосвязанного окружения на фотопроцессы с участием кислорода в газе, растворах и на поверхности фотокатализатора. Далее проанализирован прогресс в изучении супрамолекулярной фотофизики и фотохимии кислорода в Ван-дер-Ваальсовых комплексах X–O2. Дано описание метода измерения карт скоростей фотофрагментов, его возможностей, и обсуждены результаты применения этого метода для изучения супрамолекулярной фотохимии в слабосвязанных комплексах X–O2. Приведены также результаты регистрации ИК-люминесценции синглетного кислорода, образующегося при фотовозбуждении столкновительных комплексов кислорода. Рассмотрена природа электронных состояний слабосвязанных комплексов кислорода, ответственных за супрамолекулярную фотохимию в комплексах X–O2. Обсуждаются механизмы фотогенерации синглетного кислорода при возбуждении слабосвязанных комплексов X–O2.
2. СПЕКТРАЛЬНЫЕ И ФОТОХИМИЧЕСКИЕ ПРОЯВЛЕНИЯ ВЛИЯНИЯ СЛАБОСВЯЗАННОГО ОКРУЖЕНИЯ НА ФОТОПРОЦЕССЫ С УЧАСТИЕМ КИСЛОРОДА
2.1. Влияние слабосвязанного окружения на спектры поглощения кислорода
Молекулярный кислород практически прозрачен для излучения в области длин волн больше 200 нм. Несмотря на это, фотопроцессы с участием кислорода имеют первостепенную важность для химии атмосферы, процессов фотоокисления в природе, в технологической и лабораторной практике. В атмосфере Земли атомы кислорода появляются преимущественно при фотодиссоциации молекулярного кислорода O2 в результате поглощения УФ-излучения в спектральной области так называемого континуума Герцберга (200–242 нм), где энергия фотона больше энергии связи атомов в молекуле O2. Этот континуум определяется тремя переходами из основного состояния $X{}^{3}\Sigma _{g}^{ - }$ в возбужденные – $A{}^{3}\Sigma _{u}^{ + },$ $c{}^{1}\Sigma _{u}^{ - }$ и A'3Δu (рис. 1а), называемые состояниями Герцберга I, II и III соответственно. Переходы из основного в любое из этих трех состояний запрещены либо по спину, либо по орбитальной симметрии. Результатом этого запрета являются очень низкие значения сечения поглощения фотона, показанные на рис. 1б. Сечение поглощения фотона в максимуме континуума Герцберга определяется переходом в состояние Герцберга I и равно ~10–23 см2, что на 6–7 порядков величины меньше сечения для разрешенных электронных переходов. Фотохимия молекулы кислорода под действием этого излучения была рассмотрена в обзоре Паркера (Parker) [1]. Уже давно известно, что поглощение УФ-излучения драматически усиливается в результате возмущающего действия на O2 молекул окружения X при столкновениях в газовой фазе или при контакте в конденсированной среде. Вульф (Wulf) [2] и затем Финкельнбург (Finkelnburg) [3] обнаружили наличие в спектре поглощения кислорода диффузных полос в области 240–290 нм, которые перекрываются с дискретным спектром полосы Герцберга. Финкельнбург установил, что это поглощение имеет квадратичную зависимость от давления кислорода [3]. Эффект межмолекулярного взаимодействия можно рассматривать как вклад поглощения столкновительными комплексами O2–O2. Этот вклад для поглощения в континууме Герцберга в газовой фазе точно также зависит от давления и, например, для кислорода уже при атмосферном давлении он становится сопоставимым по величине с поглощением “свободных” молекул O2 [4]. На основании результатов измерений зависимости поглощения в чистом кислороде от давления Шардананд (Shardanand) оценил увеличение сечения поглощения в столкновительном комплексе O2–O2 примерно в 1000 раз по сравнению с поглощением “свободных” молекул O2 [5]. Эффект усиления поглощения как в области дискретного поглощения O2, так и в континууме Герцберга наблюдался для большого количества газов [6–11]. Сопоставление результатов измерения усиления поглощения кислорода при добавлении различных газов, представленных в работах [6–11], позволяет сделать вывод, что фактор усиления поглощения в столкновительном комплексе X–O2 может достигать величин порядка 106–107. Имеющиеся спектральные данные свидетельствуют о том, что в отличие от поглощения “изолированных” молекул O2 доминирующий вклад в усиленное поглощение вносит полоса Герцберга III, соответствующая переходу A'3Δu ← $X{}^{3}\Sigma _{g}^{ - }$ [4, 12]. В конденсированной среде, содержащей кислород, также образуются комплексы кислорода с молекулами окружения. Было показано, что они представляют собой неустойчивые короткоживущие комплексы, аналогичные столкновительным комплексам в газовой фазе [13]. При этом имеет место и усиленное поглощение в состояния Герцберга в растворах, поскольку каждая молекула растворенного O2 находится в контакте с молекулами окружения, и наличие усиления поглощения кислородом еще более заметно. Следует отметить, что спектры усиленного поглощения в слабосвязанных комплексах X–O2 по форме подобны спектру поглощения для переходов в состояния Герцберга в свободном O2 [8–10].
Рис. 1.
Потенциальные кривые для нижних электронных состояний молекулы кислорода (а) и спектр поглощения и выхода диссоциации O2 при возбуждении в континууме Герцберга (б). Приведены спектры полного поглощения, а также спектры 3-х компонент, соответствующих возбуждению состояний Герцберга I–III (нижняя панель), а также вклада колебательных компонент полосы Шумана–Рунге (верхняя панель). Рисунок 1б адаптирован из работы [1] с разрешения (2019) журнала Acc. Chem. Res.

Взаимодействие кислорода с окружением приводит не только к увеличению поглощения в полосах Герцберга, но и к появлению новых полос, отсутствующих в спектрах как кислорода, так и молекул окружения [14–18]. Впервые это наблюдал Эванс (Evans) в растворах, содержащих растворенный кислород, в работе [14]. Для ароматических [15] и ненасыщенных [16] углеводородов, содержащих кислород, было отмечено появление двух новых полос. Одна из них была отнесена к синглет-триплетному переходу T ← S в молекулах растворителя, усиленному взаимодействием с кислородом. Что касается второй, более коротковолновой и более интенсивной полосы, то Тсубомура (Tsubomura) и Малликен (Mulliken) обнаружили линейную корреляцию ее положения в спектре с потенциалом ионизации молекулы растворителя X [17]. На этом основании вторая полоса поглощения была отнесена авторами работы [17] к переходу с переносом заряда (ПЗ) в контактном комплексе:
(I)
${\text{X}}{\kern 1pt} - {\kern 1pt} {{{\text{O}}}_{2}} + h\nu \to {{{\text{X}}}^{ + }}{\kern 1pt} - {\kern 1pt} {\text{O}}_{2}^{ - }.$Такие полосы присутствовали и в криогенной кислородной матрице, содержащей молекулы алкенов [18] и ароматических углеводородов [19]. Широкое распространение это отнесение получило и потому, что наблюдающееся положение новой полосы неплохо согласуется с его простой оценкой, предложенной Бирксом (Birks) с соавт. [20]. Биркс и др. использовали для оценки энергии вертикального перехода комплекса (D–A) донора D с акцептором A в состояние с переносом заряда (${{E}_{{{\text{ПЗ}}}}}$) следующее выражение:
(1)
${{E}_{{{\text{ПЗ}}}}} = {{I}_{{\text{D}}}} - {{A}_{{\text{A}}}} - (\Delta {{E}_{E}} - \Delta {{E}_{N}}) \approx {{I}_{{\text{D}}}} - {{A}_{{\text{A}}}} - \frac{{{{e}^{2}}}}{R},$Следует отметить, что существует и другой взгляд на природу второй полосы. Хойтинк (Hoijtink) с соавторами обратили внимание на то, что эта коротковолновая полоса в системе кислород–нафталин по виду очень похожа на первую, менее интенсивную полосу, относящуюся к $T \leftarrow S$ переходу в молекулах нафталина, но по энергии сдвинута вверх примерно на 1 эВ, что соответствует энергии возбуждения молекулы O2 в синглетное состояние $\left( {{{a}^{1}}{{\Delta }_{g}}} \right)$ [21–23]. Авторы предположили, что эта коротковолновая полоса относится к кооперативному переходу с одновременным изменением электронного спина в молекулах комплекса:
(II)
$\begin{gathered} ^{1}{\text{X}}{\kern 1pt} {{ - }^{3}}{\kern 1pt} {{{\text{O}}}_{2}} + h{\nu } \to {{\,}^{3}}{\kern 1pt} {\text{X}}{\kern 1pt} - {\kern 1pt} {{{\text{O}}}_{2}}\left( {{{a}^{1}}{{\Delta }_{g}}} \right) \to \\ \to \,{{\,}^{3}}{\kern 1pt} {\text{X}} + {{{\text{O}}}_{2}}\left( {{{a}^{1}}{{\Delta }_{g}}} \right). \\ \end{gathered} $Силы осцилляторов для переходов в столкновительных комплексах, ответственных за усиление поглощения O2 в области полосы Герцберга, усиление поглощения на $T \leftarrow S$ переходах в молекулах партнера по комплексу X и для кооперативных переходов (II) также были оценены методами квантовой химии в работах Минаева с соавт. [24–26].
Хотя в большинстве цитированных выше работ появление новых полос регистрировалось при повышенном давлении кислорода, индуцированное межмолекулярным взаимодействием поглощение достаточно велико и при атмосферном давлении. На рис. 2 показаны спектры поглощения для ряда широко используемых растворителей при наличии и при отсутствии в них растворенного кислорода, заимствованные из работы [27]. Видно, что это индуцированное межмолекулярным взаимодействием поглощение как в области полос Герцберга, так и в более длинноволновой области хорошо наблюдаемо и при атмосферном давлении. То есть при воздействии УФ-излучения на эти среды необходимо учитывать наличие фотохимических процессов, обусловленных супрамолекулярным поглощением столкновительными или контактными комплексами X–O2. Естественно, возникает вопрос, а какие фотохимические процессы протекают после поглощения излучения в области этих новых полос.
Рис. 2.
а – Спектры поглощения жидких ароматических углеводородов, записанные с толщиной оптического слоя, равной 10 см. Приведены спектры насыщенных азотом бензола (A), толуола (B), о-ксилола (C) и п-ксилола (D), а также насыщенных кислородом бензола (E), толуола (F), о-ксилола (G), п-ксилола (H) и мезитилена (I). б – Спектры поглощения насыщенных азотом жидких алифатических углеводородов циклогексана (A), декалина (B) и циклооктана (D), а также насыщенных кислородом цеклогексана (E), декалина (F) и циклооктана (G) с толщиной оптического слоя 1 см. Рисунок адаптирован из работы [27] с разрешения (2019) журнала J. Phys. Chem.

2.2. Влияние слабосвязанного окружения кислорода на процессы фотоокисления
В газовой фазе фотохимические проявления столкновительно-индуцированного усиления поглощения в кислороде впервые наблюдали Слангер (Slanger) и др. [28], которые обнаружили появление озона O3 при облучении кислорода нефокусированным излучением KrF-лазера на 248 нм, что ниже порога диссоциации кислорода. Изучению этого процесса, имеющего явно выраженную зависимость от давления, была посвящена серия работ, детально обсуждаемых в обзоре Кода (Koda) и Сугимото (Sugimoto) [11]. На основании анализа результатов предыдущих исследований в [11] был сделан вывод о его механизме. Наиболее вероятным процессом, приводящим к такому образованию O3, Кода и Сугимото считают индуцированное столкновениями возбуждение молекулы кислорода в состояние Герцберга III (A'3Δu), сопровождающееся бимолекулярной реакцией:
(III)
${{{\text{O}}}_{{\text{2}}}}(A{\kern 1pt} ') + {{{\text{O}}}_{{\text{2}}}} \to {{{\text{O}}}_{{{\text{3}}~}}}\,{\text{ + }}\,\,{\text{O}}$В большом числе работ в 80-е гг. новую фотохимию растворов, содержащих растворенный кислород, изучали путем анализа продуктов фотовозбуждения в новых полосах поглощения. Не останавливаясь на всей совокупности этих исследований, рассмотрим в качестве примера полученных таким образом результатов работу Онодеры (Onodera) с соавт. [30]. При возбуждении в новых полосах поглощения, оцениваемых авторами [30] как полосы возбуждения ПЗ-состояния контактных комплексов X–O2, регистрировали продукты окисления серии замещенных ароматических соединений и полизамещенных олефинов. Для объяснения появления ряда продуктов был предложен гипотетический механизм с участием ион-радикалов ПЗ-состояния. Для замещенных нафталинов в этой работе наблюдали продукты 1,4-присоединения кислорода, что совпадает с продуктами реакций с участием синглетного кислорода 1O2. На этом основании был сделан вывод о генерации 1O2 в этих экспериментах. Для ряда аминов в работе [30] химических изменений не обнаружено. Это было объяснено тем обстоятельством, что вышеупомянутые соединения не реагируют с 1O2 или являются его эффективными тушителями. Аналогичные исследования фотоокисления углеводородов при возбуждении новых полос проводили в криогенной кислородной матрице [18, 19]. Наблюдалось окисление углеводородов, причем спектральный порог для фотоокисления многих молекул соответствовал энергии фотона, существенно меньшей, чем энергия разрыва O–O-связи в кислороде. Для различных алкенов регистрировались различные каналы фотоокисления: образование аддукта кислорода, цис–транс-изомеризация, разрыв двойной связи с формированием двух карбонильных соединений, появление CO, CO2, O3 и HO2. Все наблюдаемые процессы также были интерпретированы как результат возбуждения ПЗ-состояния в контактных комплексах X–O2.
Появление синглетного кислорода ${{{\text{O}}}_{2}}\left( {{{a}^{1}}{{\Delta }_{g}}} \right)$ при возбуждении полос супрамолекулярного поглощения контактных комплексов X–O2 было зарегистрировано и напрямую. Огилби (Ogilby) с соавторами [27, 31–33] и Дарманян с Шариповым [34] установили по характерной люминесценции в ближнем ИК-диапазоне, что при возбуждении растворителей, содержащих кислород, в коротковолновой полосе супрамолекулярного поглощения образуется синглетный кислород ${{{\text{O}}}_{2}}\left( {{{a}^{1}}{{\Delta }_{g}}} \right).$ Опираясь на отнесение Тсубомуры и Малликена [17], авторы вышеуказанных работ сделали вывод, что образование синглетного кислорода обусловлено возбуждением контактных комплексов X–O2 в состояние с переносом заряда. Синглетный кислород ${{{\text{O}}}_{2}}\left( {{{a}^{1}}{{\Delta }_{g}}} \right)$ является первым возбужденным состоянием O2 (см. рис. 1а) и представляет собой реакционноспособную частицу, играющую важную роль в процессах фотоокисления [35]. Синглетный кислород – важный участник многих биохимических процессов, а также ключевой агент в фотодинамической терапии [36]. Это вызывает дополнительный интерес к механизму его образования при возбуждении слабосвязанных комплексов кислорода.
Другим примером драматического влияния окружения на фотопроцессы с участием кислорода является селективное фотоокисление кислородом молекул углеводородов в порах цеолитов, которое наблюдается под действием излучения видимого диапазона, характеризующегося малой энергией фотона (hν ≤ 3 эВ) [37]. Так, при воздействии излучения видимого диапазона на смесь легких насыщенных или ненасыщенных углеводородов с кислородом в нанометровых порах цеолитов протекает высокоселективное фотоокисление с образованием альдегидов или кетонов [38–40] или гидроперекисей [41]. Высокий квантовый выход в сочетании с доступной высокой интенсивностью ламп видимого диапазона делают такой фотокатализ практически значимым [40]. Следует напомнить, что ни кислород, ни эти углеводороды не имеют полос поглощения в видимом диапазоне. Появляющееся поглощение связывают с участием ПЗ-состояний контактных комплексов X–O2 (где X – углеводороды) [37]. То, что в этих системах уже энергия кванта видимого излучения достаточна для возбуждения состояния с переносом заряда, объясняется существенным понижением энергии ПЗ-состояния ${{{\text{X}}}^{ + }}{\kern 1pt} - {\kern 1pt} {\text{O}}_{2}^{ - }$ на поверхности цеолита вследствие стабилизации его кулоновским взаимодействием с ионами поверхности цеолита (Ca2+ и др.) [38, 40–44]. Оцениваемая энергия стабилизации составляет около 2 эВ, что полагается достаточным для сдвига спектра поглощения в ПЗ-состояние комплекса из УФ- в видимый диапазон [41]. Есть и другая версия, основанная на результатах квантово-химического исследования, которая объясняет влияние поверхности локализацией пары X–O2 в конфигурации, благоприятной для возбуждения ПЗ-состояния [45]. Что касается механизма такого каталитического фотоокисления, то на первых стадиях предполагается перенос протона в ПЗ-состоянии с катион-радикала углеводорода на супероксиданион с появлением радикала HO2 и дальнейшим образованием гидроперекиси [41]. Формирование карбонильных соединений вероятно протекает в результате распада диоксетана, появляющегося в результате прямого присоединения супероксиданиона к катион-радикалу углеводорода [41]. Экспериментальное исследование механизма каталитического фотоокисления внутри цеолитовых пор является сложной задачей. Для изучения влияния слабосвязанного окружения на фотопроцессы с участием кислорода необходима более простая модельная система.
Достигнутый к настоящему времени прогресс в понимании природы рассмотренных выше процессов супрамолекулярной фотохимии слабосвязанных комплексов кислорода связан с применением в качестве модельной системы Ван-дер-Ваальсовых комплексов X–O2.
3. СУПРАМОЛЕКУЛЯРНАЯ ФОТОХИМИЯ ВАН-ДЕР-ВААЛЬСОВЫХ КОМПЛЕКСОВ X–O2
3.1. Ван-дер-Ваальсов комплекс X–O2 как модельная система. Первые эксперименты
Поскольку первичный агент супрамолекулярной фотохимии – это столкновительный или контактный комплекс X–O2, то естественной является постановка задачи исследования фотоиндуцированных процессов в таком комплексе. Это оказалось возможным реализовать при использовании в качестве модельной системы Ван-дер-Ваальсовых комплексов кислорода X–O2. Ван-дер-Ваальсовы комплексы молекул можно генерировать в высокой концентрации с помощью техники импульсных молекулярных пучков [46, 47]. Энергия Ван-дер-Ваальсова взаимодействия обычно не превышает 3 ккал/моль. Комплексы с такой энергией связи становятся устойчивыми только при очень низкой температуре. Вышеуказанные комплексы генерируют при расширении газа в вакуум через сверхзвуковое сопло, что позволяет существенно охлаждать, в принципе до единиц К, температуру поступательных, вращательных и колебательных степеней свободы. При расширении газовой смеси, содержащей молекулы X и O2 в газе-носителе M (часто используется гелий), происходит образование Ван-дер-Ваальсовых комплексов в результате столкновительной рекомбинации:
(IV)
${\text{X\;}} + {{{\text{O}}}_{2}}{\text{\;}} + {\text{M}} \to {\text{X}}{\kern 1pt} - {\kern 1pt} {{{\text{O}}}_{2}}{\text{\;}} + {\text{M}}{\text{.}}$Далее при расширении формируется молекулярный пучок, содержащий холодные комплексы. Благодаря бесстолкновительным условиям молекулярного пучка для исследования процесса фотодиссоциации Ван-дер-Ваальсовых комплексов возможно привлечение методов, разработанных для детального изучения диссоциции молекул. Более того, Ван-дер-Ваальсов комплекс имеет фиксированную структуру, которая может быть рассчитана методами квантовой химии. Это дает надежду на более глубокое понимание процессов в Ван-дер-Ваальсовых комплексах. ДеБоер (DeBoer) и Янг (Young) использовали технику импульсных молекулярных пучков для генерации Ван-дер-Ваальсовых комплексов кислорода с бензолом C6H6–O2 и изучения их фотодиссоциации при возбуждении в области полосы Герцберга лазерным излучением с длиной волны около 226 нм [48]. При этом ДеБоер и Янг регистрировали образующиеся при фотодиссоциации атомы кислорода (3PJ):
(V)
${\text{X}}{\kern 1pt} - {\kern 1pt} {{{\text{O}}}_{2}}{\text{\;}} + h{\nu } \to {\text{O}}\left( {^{3}{{P}_{J}}} \right) + \,\,~ \ldots $Рис. 3.
Схема резонансно-усиленной многофотонной ионизации РУМФИ (2 + 1) атомов кислорода в состоянии ${\text{O}}{{(}^{3}}{{P}_{J}}){\text{.}}$

Очень значительный прогресс в понимании супрамолекулярной фотохимии слабосвязанного кислорода достигнут в последние 12 лет, и связано это, в первую очередь, с использованием техники измерения карт скоростей фотофрагментов для изучения фотодиссоциации Ван-дер-Ваальсовых комплексов X. В следующей главе приведено описание этой техники.
3.2. Техника измерения карт скоростей фотофрагментов
Техника ионного имиджинга (ion imaging), позволяющая измерять распределение ионов по скоростям, впервые была реализована в работе Хьюстона (Houston) и Чандлера (Chandler) [52]. Позднее Эппинком (Eppink) и Паркером (Parker) был реализован ее усовершенствованный вариант (velocity map imaging), который позволил существенно поднять разрешение метода [53]. Этот вариант имиджинга очень широко используется для изучения фотохимических процессов в молекулах и молекулярных комплексах [54–56]. Все аспекты этой техники детально описаны в монографии [54], а ее современное состояние обсуждается в работе [56]. Ниже мы рассмотрим основные особенности этого метода и возможности, которые он дает при исследовании фотохимических процессов.
На рис. 4 приведена типичная схема экспериментальной установки для измерения карт скоростей фотофрагментов. Газовая смесь X + O2 в избытке газа-носителя (обычно He) через импульсный газовый клапан поступает в вакуумную камеру. При расширении в вакуум образуется сверхзвуковая струя, в которой происходит охлаждение газа и образование Ван-дер-Ваальсовых комплексов. При дальнейшем расширении с помощью скиммера формируется молекулярный пучок. Этот пучок, содержащий молекулы и молекулярные комплексы, распространяется по оси спектрометра. Через отверстие в первом электроде частицы пучка попадают в промежуток между первым и вторым электродами (см. рис. 4), где происходит возбуждение молекул или комплексов сфокусированным лазерным излучением, которое поляризовано перпендикулярно оси спектрометра. Рассмотрим основные особенности метода на примере измерения карты скоростей для атомов кислорода, образующихся при фотодиссоциации молекулы O2:
(VI)
${{{\text{O}}}_{2}} + h{\nu } \to {\text{O}}{{(}^{3}}{{P}_{J}}) + {\text{O}}{{(}^{3}}{{P}_{J}}).$Рис. 4.
Схема экспериментальной установки, используемой для измерения карт скоростей фотоионов в стандартной схеме (постоянное напряжение на электродах) и в варианте “слайсинга” (импульсное напряжение на электродах) ионного облака фотофрагментов.

При поглощении фотона с энергией $h\nu {\text{,}}$ превышающей энергию связи ${{D}_{0}}$(О–О) 5.117 эВ (λvac < 242.3 нм), молекула ${{{\text{O}}}_{2}}$ распадается с образованием двух атомов кислорода ${\text{O}}{{(}^{3}}{{P}_{J}}).$ Указанная энергия связи соответствует диссоциации на два атома в основном состоянии O(3P2). Состояния атома кислорода O(3P1) и O(3P0) лежат выше основного состояния по энергии на 0.020 и 0.028 эВ соответственно. Атомы кислорода разлетаются по отношению к центру масс ${{{\text{O}}}_{2}}$ с кинетической энергией Et, равной
(2)
${{E}_{{\text{t}}}} = \frac{1}{2}\left( {h\nu - {{D}_{0}}\left( {{\text{O}}{\kern 1pt} - {\kern 1pt} {\text{O}}} \right) - {{E}_{{{\text{int}}}}}} \right),$Рис. 5.
Имиджи атомов кислорода O(3P2), образующихся при фотодиссоциации молекулы O2 под действием лазерного излучения с длиной волны 225.656 нм: а – 2D-имидж: б – слайс 2D-имиджа; в – распределение по кинетической энергии атомов, построенное по результатам инверсии 2D-имиджа (а) с помощью преобразования Абеля. Стрелкой показано направление поляризации лазерного излучения.

Рис. 6.
Имиджи атомов кислорода O(3P2), образующихся при УФ-фотодиссоциации молекулы кислорода (а), а также Ван-дер-Ваальсовых комплексов X–O2 (б–д) излучением с λvac = 225.656 нм, обеспечивающим фотодиссоциацию комплексов и резонансную фотоионизацию РУМФИ (2 + 1) атомов O(3P2). В каждой паре имиджей левый соответствует стандартному 2D-изображению (2D-имидж), а правый – слайсу центральной части ионного облака. Двойная стрелка показывает направление поляризации возбуждающего излучения; 0–5 – номера каналов. Имиджи заимствованы из работы [64] с разрешения журнала J. Chem. Phys.

Измерение кинетической энергии фотофрагментов в сочетании с их идентификацией по РУМФИ-спектрам позволяет с учетом энергетического баланса (уравнение (2) для фотодиссоциации кислорода) идентифицировать частицы-предшественники, установить таким образом частицы и их электронные состояния, из которых образовались регистрируемые фотофрагменты, и, соответственно, получить информацию о механизме изучаемого фотопроцесса. Следует отметить успешное применение этого метода для измерения энергии связи в молекулах и радикалах [58–60].
Имидж для ионов O+, показанный на рис. 5а и 5б, также демонстрирует угловую асимметрию распределения по направлениям вылета атомов O по отношению к поляризации излучения (вектору электрического поля световой волны ${{\vec {E}}_{0}}$). Видно, что сигнал выше для атомов, вылетающих в направлениях полюсов (в направлении поляризации излучения). Это распределение определяется симметрией волновой функции начального и конечного электронных состояний возбуждаемого перехода, а точнее направлением вектора дипольного момента перехода ($\vec {d}$) по отношению к направлению оси молекулы. Допустим, что возбуждается двухатомная молекула и что возбуждаемый электро-дипольный переход является “параллельным”, то есть направление вектора дипольного момента перехода ($\vec {d}$) совпадает с направлением оси молекулы. В этом случае вероятность поглотить квант максимальна для молекул, ориентированных по полю ${{\vec {E}}_{0}}.$ Зависимость этой вероятности P(θ) от угла θ между направлениями ${{\vec {E}}_{0}}$ и осью молекулы для “параллельного” перехода может быть представлена как P(θ) ∝ cos2θ. Полагаем, что энергия поглощенного кванта превышает энергию связи в молекуле. Если время жизни возбужденной молекулы много меньше периода ее вращения, то поглотившая квант молекула диссоциирует до того, как изменится ее ориентация в пространстве. А это значит, что функция распределения f(θ) фрагментов диссоциации (атомов в данном случае) по углам разлета (в расчете на единичный телесный угол) по отношению к ${{\vec {E}}_{0}}$ будет повторять угловую зависимость вероятности поглощения фотона P(θ), т.е. f(θ) ∝ cos2θ. Если $\vec {d}$ перпиндикулярно оси молекулы (переход ”перпендикулярный”), то f(θ) ∝ sin2θ. В общем случае произвольной поляризации перехода, характеризуемой параметром анизотропии β, эта функция распределения определяется выражением:
(3)
$f(\theta ) = {{({\text{4}}\pi )}^{{ - {\text{1}}}}}[{\text{1}} + \beta {{P}_{{\text{2}}}}({\text{cos}}\theta )],$Для улучшения разрешения карт скоростей используются также модификации техники измерения карт скоростей. В обзорных работах [53–55] подробно рассмотрены различные вариации метода, позволяющие “вырезание” (слайсинг) узкого центрального слоя из облака ионов, образующегося при фотоионизации фрагментов диссоциации. Вкратце рассмотрим один из вариантов, основанный на использовании “задержки вытягивания” (delayed extraction) ионов из области фотоионизации [63] и применявшийся в экспериментах с комплексами кислорода, которые будут обсуждаться в дальнейшем. В этом подходе фотогенерация фрагментов и их фотоионизация осуществляется при отсутствии электрического поля между электродами масс-спектрометра. Вытягивающее и ускоряющее поля включаются (см. рис. 4) с задержкой после лазерного импульса, регулируемой в диапазоне 100–2000 нс, в течение которой облако разлетающихся фотоионов расширяется. После включения вытягивающего напряжения ионы двигаются к детектору. Обусловленный разлетом ионов во время задержки относительно большой объем облака уже не обеспечивает условий временнóй фокусировки ионов одной массы на детекторе, характерной для время-пролетного масс-спектрометра. При этом облако ионов одной массы прилетает на микроканальную пластину (МКП) детектора в течение интервала времени большего, чем в отсутствие указанной задержки. Далее, если открывать детектор достаточно коротким высоковольтным импульсом (рис. 4), то можно “вырезать” центральную часть облака ионов, что и позволяет увеличить разрешение его изображения. Результат слайсинга ионного облака атомов кислорода показан на рис. 5б.
3.3. Карты скоростей атомов кислорода O(3PJ)
К настоящему времени были измерены карты скоростей атомов кислорода O(3P2), образующихся при фотодиссоциации Ван-дер-Ваальсовых комплексов кислорода X–O2, где партнерами Х по комплексу были: ксенон Xe, циклогексан C6H12, этилен C2H4, пропилен C3H6, бутадиен C4H6, изопрен C5H8, йодистый метил CH3I (CD3I), бензол C6H6 и вода H2O [64–68]. Бóльшая часть измерений карт скоростей атомов O была выполнена в однолазерных экспериментах, где излучение лазера (λvac ≈ 226 нм)) использовали одновременно для возбуждения комплекса X–O2 и резонансной фотоионизации РУМФИ (2 + 1) образующихся атомов O. Ниже на рис. 6 в качестве примера результатов таких измерений приведены имиджи для фотодисоциации 4-х комплексов X–O2 (X = Xe, C6H12, C3H6, CH3I) в сравнении с фотодиссоциацией свободного O2 [64]. При этом для комплексов X–O2 (X = C3H6, CH3I) в имидже наблюдался еще и канал с заметно большей кинетической энергией атомов, показанный на рис. 7. Результаты для комплексов драматически различны как по интенсивности сигнала атомов O, так и по виду имиджа там, где партнером по комплексу является молекула. Это значит, что фотодиссоциация кислорода в слабосвязанном комплексе радикально отличается от фотодиссоциации несвязанного O2. В полученных имиджах можно выделить вклад 5 дополнительных каналов фотодиссоциации, различающихся по кинетической энергии и угловому распределению атомов, которые показаны на рис. 6 вверху справа и пронумерованы. Все пронумерованные каналы в дальнейшем мы будем обозначать как К0–К5, при этом канал К0 соответствует образованию атомов при фотодиссоциации свободных молекул O2.
Рис. 7.
Имидж для атомов O(3PJ), образующихся при фотодиссоциации Ван-дер-Ваальсовых комплексов: а – CH3I–O2 с регистрацией O(3P2); б – CH3I–O2 с регистрацией O(3P0) и в – C3H6–O2 с регистрацией O(3P2). Имиджи (а) и (в) соответствуют имиджам на рис. 6д и 6г, но для них показано и внешнее кольцо, искусственно усиленное в 5–10 раз для лучшей видимости на рисунке. Имиджи заимствованы из работы [64] с разрешения журнала J. Chem. Phys.

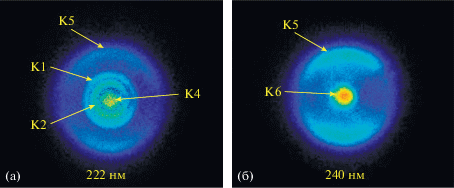
В работе [66] была измерена зависимость карт скоростей от длины волны возбуждающего излучения. В этих экспериментах использовали два лазера. Длину волны одного лазера варьировали, а длина волны второго была фиксированной и позволяла резонансно фотоионизовать образующиеся атомы O(3PJ). На рис. 8 представлены имиджи для комплекса изопрена с кислородом C5H8–O2 на двух длинах волн 222.0 ($h{\nu } = 5.59~\,\,{\text{эВ}}$) и 240.0 нм ($h{\nu } = 5.17\,\,{\text{эВ}}$). При изменении энергии фотона возбуждающего излучения положение колец, соответствующих различным каналам, смещается по энергии. Это также помогает понять происхождение наблюдаемых каналов фотодиссоциации комплексов. Далее последовательно будут рассмотрены каналы фотодиссоциации, их механизм и природа соответствующих возбужденных состояний комплексов X–O2.
Рис. 8.
Имиджи для атомов O(3P0), образующихся при фотодиссоциации Ван-дер-Ваальсовых комплексов изопрена с кислородом C5H8–O2. Возбуждение осуществляли излучением с длиной волны 222.0 (а) и 240.0 нм (б). Для фотоионизации O(3P0) использовали излучение второго лазера с фиксированной длиной волны 226.233 нм. При этом из имиджа был вычтен вклад атомов O(3P0), обусловленный возбуждением комплексов на длине волны зондирующего излучения (226.233 нм). Имиджи заимствованы из работы [66] с разрешения журнала J. Chem. Phys.
3.4. Природа возбужденных состояний и механизмы супрамолекулярной фотохимии комплексов X–O2
Возбуждение “возмущенного” состояния Герцберга. Канал К1. Для всех без исключения комплексов наблюдается канал с энергией немного меньшей, чем при фотодиссоциации свободного O2. На рис. 9 в качестве примера показано распределение по энергии атомов O(3P2), полученное в результате инверсии 2D-имиджей с рис. 7а для фотодиссоциации O2 и с рис. 7д для фотодиссоциации CH3I–O2. Наличие угловой анизотропии (β = –0.48) указывает на то, что O2 в возбужденном состоянии комплекса очень быстро распадается с характерным временем короче периода вращения. Это свидетельствует о том, что молекула O2 оказывается в ковалентном возбужденном состоянии. По величине сигнала это основной канал образования атомов кислорода, поэтому можно сделать вывод, что именно он определяет усиление поглощения в слабосвязанных комплексах X–O2. Этот канал отнесен к процессу возбуждения
(VII)
$\begin{gathered} {\text{X}}{\kern 1pt} - {\kern 1pt} {{{\text{O}}}_{2}}\left( {^{3}{\Sigma }_{g}^{ - }} \right) + h\nu \to \\ \to {\text{X}}{\kern 1pt} - {\kern 1pt} {{{\text{O}}}_{2}}\left( {A{\kern 1pt} '{{\,}^{3}}{{\Delta }_{u}}} \right) \to {\text{X}} + {\text{O}} + {\text{O,}} \\ \end{gathered} $Рис. 9.
Распределение по энергии атомов кислорода O(3P2), образующихся при фотодиссоциации свободных молекул O2 и комплексов CH3I–O2. Распределение получено в результате инверсии 2D-имиджей, представленных на рис. 6а и 6д соответственно. Также показано значение параметра анизотропии для образующихся из комплекса атомов.

Рис. 10.
Схема уровней электронных состояний комплекса X–O2, определяющих “усиление” поглощения на переходе, запрещенном в свободном O2.

Блок и др. [6] рассматривали в качестве ${{{\varphi }}_{2}}$ состояние с переносом заряда (ПЗ) комплекса Х+–О2–. Малликен, анализируя переходы в ПЗ-состояние комплексов X–Y2, сделал вывод, что матричный элемент перехода должен совпадать по направлению с постоянным дипольным моментом состояния ПЗ (${{\vec {d}}_{{{\text{ПЗ}}}}}$) [70], направленным из центра ${\text{O}}_{2}^{ - }$ на катион ${{{\text{X}}}^{ + }}{\text{.}}$ На рис. 11 показано направление разлета атомов кислорода при ориентации матричного элемента перехода под углом θ по отношению к направлению поляризации возбуждающего излучения. Максимум вероятности поглощения реализуется при таком положении комплекса, когда ${{\vec {d}}_{{{\text{ПЗ}}}}}$ направлен параллельно поляризации излучения (θ = 0). Если комплекс имеет Т-образную структуру, как показано на рис. 11, то атомы кислорода будут разлетаться преимущественно перпендикулярно направлению поляризации излучения, что и наблюдалось в эксперименте. На рис. 12 приведена рассчитанная ab initio структура комплекса CH3I–O2 [65]. В состоянии ПЗ комплекса положительный заряд локализован преимущественно на атоме иода, поэтому постоянный дипольный момент ${{\vec {d}}_{{{\text{ПЗ}}}}}$ направлен из центра ${\text{O}}_{2}^{ - }$ на катион ${{{\text{I}}}^{ + }}{\text{.}}$ Результаты расчета, представленные на рис. 12, демонстрируют, что ось молекулы O2 в комплексе близка к перпендикулярному положению по отношению к направлению ${{\vec {d}}_{{{\text{ПЗ}}}}}.$ Т-образная структура в качестве основной получена также и для комплексов с X = Xe, C6H12, C3H6 [65]. Для комплексов Xe–O2 этот вывод подтверждается данными эксперимента [71]. Таким образом, наблюдаемая угловая анизотропия разлета атомов кислорода соответствует тому, что в комплексе к состоянию Герцберга III молекулы O2 примешивается волновая функция состояния с переносом заряда, что и приводит к усилению поглощения в слабосвязанных комплексах кислорода. Рассмотрение симметрии состояний Герцберга и ПЗ также показало, что следует ожидать эффективного смешения ПЗ-состояния только с состоянием Герцберга III [64]. Необходимо отметить, что подтверждаемый экспериментом механизм усиления поглощения не предполагает прямого возбуждения в ПЗ-состояние комплекса, которое в исследованных комплексах лежит по энергии существенно выше исследуемой спектральной области. Речь идет о небольшой “примеси” волновой функции ПЗ-состояния. В первом порядке теории возмущений [72] “примесь” волновой функции ПЗ-состояния обратно пропорциональна $a\sim \frac{1}{{\Delta E}}$ разности энергии “невозмущенного” ПЗ-состояния ${{{\text{X}}}^{ + }}{\kern 1pt} - {\kern 1pt} {\text{O}}_{2}^{ - }$ (${{{\varphi }}_{2}}$) и состояния Х–О2${{(}^{3}}{{\Delta }_{u}})$ (${{{\varphi }}_{1}}$) комплекса, представленных на рис. 10. Вероятность перехода, показанного на рис. 10, из основного в “возмущенное” состояние ${{{\Psi }}_{1}}$ пропорциональна квадрату “примеси” ПЗ-состояния $a\sim \frac{1}{{{{{\left( {\Delta E} \right)}}^{2}}}}.$ Следует отметить, что Блок и др. [6] применяли такую модель и получили хорошее согласие при описании с использованием уравнения (1) экспериментально наблюдаемой нелинейной зависимости эффективности столкновительного усиления поглощения в кислороде в газовой фазе от потенциала ионизации молекул партнера по столкновениям X.
Рис. 11.
Схема фотодиссоциации комплекса X–O2 с Т-образной структурой в результате возбуждения в состояние Герцберга III с “примесью” ПЗ-состояния (канал К1).

Рис. 12.
Рассчитанная ab initio структура и энергия связи (${{E}_{{{\text{bind}}}}}$) в трех самых устойчивых конфигурациях Ван-дер-Ваальсова комплекса CH3I–O2. Рисунок из работы [65] адаптирован с разрешения журнала J. Chem. Phys.

Измерение энергии Ван-дер-Ваальсовой связи в комплексах кислорода X–O2. Отмеченное выше снижение кинетической энергии атомов O(3PJ), образующихся при фотодиссоциации комплексов X–O2, по сравнению с фотодиссоциацией свободной молекулы O2 можно использовать для измерения энергии Ван-дер-Ваальсовой связи. Это было реализовано в работе [65]. В табл. 1 приведены полученные с помощью измеренных карт скоростей значения энергии Ван-дер-Ваальсовой связи ${{E}_{{{\text{bind}}}}}.$ Ван-дер-Ваальсовы взаимодействия играют критически важную роль в биологических системах. Для определения структуры молекулярных комплексов и изучения динамики структурных изменений методами молекулярной динамики необходимо знание энергетических параметров межмолекулярного взаимодействия. В настоящее время усилия большого числа исследователей направлены на развитие квантово-химических подходов для расчета таких параметров [73]. Для верификации разрабатываемых методов необходимы экспериментальные данные, которые очень ограничены. К настоящему времени эти данные получают преимущественно с помощью спектроскопии высокого разрешения [74]. Реализованный в работе [65] новый подход для измерения энергии Ван-дер-Ваальсовой связи, основанный на измерении карт скоростей фрагментов фотодиссоциации комплексов, дает инструмент для получения таких экспериментальных данных. Этот новый метод, использованный для комплексов кислорода, может быть применен и для слабосвязанных комплексов других молекул.
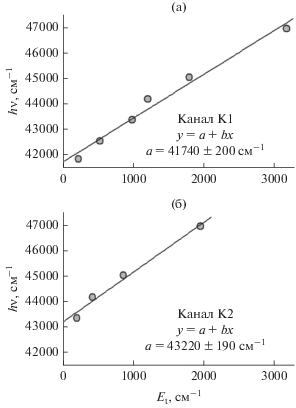
Кооперативное электронно-колебательное возбуждение комплекса. Канал К2. На имиджах для атомов O, образующихся при фотодиссоциации комплексов C5H8–O2 (рис. 8 а) и CH3I–O2 (рис. 6д), виден еще один канал (К2) с меньшей энергией, чем для К1, и таким же распределением по направлениям вылета. Подобие углового распределения указывает на то, что этот канал также связан с возбуждением O2 в комплексе в состояние Герцберга III. Более низкую энергию атомов O можно объяснить тем, что при возбуждении в канале К2 меняется состояние обеих молекул комплекса, а именно: одновременно с электронным возбуждением O2 происходит колебательное возбуждение молекулы X. По-видимому, для этого процесса есть аналогия с наблюдавшимся ранее Чоу (Chou) и Ханом (Khan) процессом люминесценции синглетного кислорода в алкилгалогенидах R3C–Hal (Hal = F, Cl, Br и I) в качестве растворителей [75]. Авторы обнаружили наряду с обычной люминесценцией синглетного кислорода на 1.27 мкм также слабую люминесценцию в длинноволновой области, отличающуюся по энергии фотона на энергию кванта колебания C–Hal молекулы растворителя. Минаев объяснил это явление тем, что фосфоресценция синглетного кислорода индуцируется межмолекулярным взаимодействием синглетного кислорода с молекулами растворителя, очень чувствительным к расстоянию между молекулами [76]. В модели Минаева молекула растворителя ориентирована атомом галогена на синглетный кислород. При этом колебание С–Hal модулирует межмолекулярное расстояние и, соответственно, величину межмолекулярного взаимодействия, ответственного за усиление фосфоресценции синглетного кислорода. Природу колебания, которое возбуждается в молекуле X, можно определить по зависимости кинетической энергии фотофрагментов, образующихся в каналах К1 и К2, от энергии возбуждающего фотона. На рис. 13 показана такая зависимость для комплекса C5H8–O2 из работы [66]. Разница величин энергии фотона, полученных при экстраполяции зависимостей на нулевое значение кинетической энергии атомов О, составляет около 1500 см–1, что для молекулы изопрена соответствует частоте валентного C=C-колебания [77]. В возбужденном состоянии комплекса CH3I–O2 расстояние I–O2 очень влияет на “примесь” (вклад) ПЗ-состояния, поскольку его энергия сильно зависит от расстояния X–O2. Поэтому колебание, модулирующее I–O2, участвует в переходе. Сдвиг по полной кинетической энергии атомов O для канала К2 по отношению к К1 составляет около 1100 см–1, что примерно соответствует энергии двух квантов C–I-колебания молекулы CH3I [77].
Рис. 13.
Зависимость кинетической энергии Et атомов O(3P0), образующихся в каналах К1 (а) и К2 (б), от энергии возбуждающего фотона для комплекса C5H8–O2. Рисунок из работы [66] адаптирован с разрешения журнала J. Chem. Phys.
Возбуждение вблизи порога диссоциации. Канал К6. При уменьшении энергии фотона возбуждающего излучения энергия атомов O, соответствующая каналу К1, снижается, что видно на имиджах рис. 8 для комплекса C5H8–O2. На рис. 14 приведен интегральный сигнал для канала К1 в зависимости от длины волны возбуждающего излучения. Удивительно наличие распада кислорода в комплексе при энергии фотона ниже порога фотодиссоциации свободной молекулы кислорода, также отмеченного на рис. 14. Это позволяет нам предположить, что вблизи порога либо протекает процесс с образованием связи C5H8–O:
(VIII)
${{{\text{С}}}_{{\text{5}}}}{{{\text{Н}}}_{{\text{8}}}}{\kern 1pt} --{\kern 1pt} {{{\text{О}}}_{{\text{2}}}} + h\nu \to {\text{O}}{{(}^{3}}{{P}_{0}}{\text{)}}\,\,{\text{ + }}\,\,{{{\text{C}}}_{5}}{{{\text{H}}}_{8}}{\kern 1pt} --{\kern 1pt} {\text{O,}}$(IX)
${\text{(}}{{{\text{С}}}_{{\text{5}}}}{{{\text{Н}}}_{{\text{8}}}}{\kern 1pt} --{\kern 1pt} {{{\text{О}}}_{{\text{2}}}}){\kern 1pt} * + \,\,h\nu \to {\text{O}}{{(}^{3}}{{P}_{0}}{\text{)}}\,\,{\text{ + }}\,\,{\text{O}}{{{\text{(}}}^{3}}{{P}_{J}}{\text{) + }}{{{\text{С}}}_{{\text{5}}}}{{{\text{Н}}}_{{\text{8}}}}{\text{.}}$Рис. 14.
Интегральный сигнал канала К1 в имидже атомов O(3P0), образующихся при фотодиссоциации комплексов C5H8–O2 (◼), в зависимости от длины волны возбуждающего излучения [66]. Символы (⚪) соответствуют сигналу от атомов кислорода при фотовозбуждении несвязанного кислорода [66]. Рисунок адаптирован из работы [66] с разрешения журнала J. Chem. Phys.

Аналогичные механизмы были предположены Кода и Сугимото для объяснения фотогенерации озона при возбуждении кислорода ниже порога диссоциации [11].
Возбуждение комплекса с одновременным изменением электронного спина молекул-партнеров (“двойной спин-флип”). Канал К5. Регистрация синглетного кислорода ${{O}_{2}}(a{{\,}^{1}}{{\Delta }_{g}}).$ Самую высокую кинетическую энергию имеют атомы O(3P0), образующиеся в канале К5, показанном на рис. 7 и 8 . В однолазерном эксперименте с длиной волны возбуждающего излучения λvac = 226.233 нм (hν = 5.48 эВ) кинетическая энергия атомов O(3P0) была равна ~0.7 эВ. Этот канал наблюдался для ряда комплексов X–O2 (X = C2H4, C3H6, C4H6, C5H8, CH3I, CD3I, C6H6) [64–67]. Поскольку энергия атомов O(3P0) для канала К5 в первом приближении не зависела от партнера по комплексу, то был сделан вывод, что вышеуказанный канал соответствует фотодиссоциации кислорода [64]. При этом суммарная кинетическая энергия двух атомов O составляла 1.4 эВ. Авторы работы [64], в которой канал К5 впервые наблюдался в качестве источника атомов кислорода с такой энергией, предположили, что атомы кислорода образуются в результате фотодиссоциации супероксиданиона:
Но в работе [66] при изучении комплексов изопрена C5H8–O2, было показано, что при фотодиссоциации комплекса у процесса К5 нет ионных предшественников. Был предложен новый механизм появления канала К5, связанный с фотодиссоциацией молекулы кислорода в синглетном состоянии ${{{\text{O}}}_{2}}(a{{\,}^{1}}{{\Delta }_{g}})$ фотоном того же лазерного импульса, который возбуждает комплекс X–O2:
(X)
$\begin{gathered} {{{\text{O}}}_{2}}({{a}^{{\,1}}}{{\Delta }_{g}}) + h{\nu }\left( {5.48\,\,{\text{эВ}}} \right) \to {\text{O}}{{(}^{3}}{{P}_{0}}) + {\text{O}}{{(}^{3}}{{P}_{2}}), \\ {{E}_{{{\text{порог}}}}} = 4.168\,\,{\text{эВ}}. \\ \end{gathered} $Для этого процесса избыточная энергия hν – $ - \,\,{{E}_{{{\text{порог}}}}} = 1.312$ эВ лишь незначительно меньше суммарной кинетической энергии образовавшихся атомов. Это указывает, что синглетный кислород образуется колебательно холодным. В работе [78] была реализована фотодиссоциация синглетного кислорода ${{{\text{O}}}_{2}}({{a}^{{\,\,1}}}{{\Delta }_{g}}),$ который генерировался при формировании молекулярного пучка в результате прохождения расширяющейся газовой струи через зону электрического разряда. В продуктах фотодиссоциации регистрировали атомы O(3P0) с той же кинетической энергией и тем же угловым распределением, что и наблюдаемые для канала К5 в работах [64–67] и показанные на рис. 7 и 8 . Это подтверждает вывод об образовании синглетного кислорода при фотодиссоциации комплексов X–O2. Недавно в работе Богомолова с соавт. [68] была реализована новая схема резонансно-усиленной многофотонной ионизации РУМФИ [(1 + 1') + 1] молекулярного кислорода (рис. 15), которая позволила напрямую зарегистрировать состояния молекулярного кислорода O2, возникающие в результате возбуждения комплекса изопрена с кислородом C5H8–O2. В этой схеме одновременно используются перестраиваемое излучение лазера на красителе видимого диапазона и его вторая гармоника (УФ-излучение), что дает возможность, используя один импульсный лазер на красителе, и возбуждать комплекс X–O2 (УФ-излучение), и фотоионизовать затем молекулы кислорода как в основном, так и в возбужденном синглетном состоянии ${{{\text{O}}}_{2}}(a{{\,}^{1}}{{\Delta }_{g}}),$ а также измерять карты скоростей этих молекул. На рис. 16 показан полученный спектр сигнала РУМФИ молекул O2. На рис. 16а представлены экспериментальные спектры, зарегистрированные при фотодиссоциации комплексов, а на рис. 16б – пересчитанные на используемую схему фотоионизации РУМФИ [(1 + 1') + 1] литературные спектры кислорода в основном ${}_{{}}^{3}{\Sigma }_{g}^{ - }$ и возбужденном синглетном $a{{\,}^{1}}{{\Delta }_{g}}$ состояниях, полученные при другом способе генерации. В регистрируемом спектре на рис. 16а наблюдается группа линий в области около 467–469 нм, по положению соответствующая спектру РУМФИ [(1 + 1') + 1] кислорода в синглетном состоянии ${{{\text{O}}}_{2}}({{a}^{{\,\,1}}}{{\Delta }_{g}})$, показанному ниже на рис. 16б. Это подтверждает вывод об образовании синглетного кислорода ${{{\text{O}}}_{2}}({{a}^{{\,\,1}}}{{\Delta }_{g}})$ при УФ-фотодиссоциации комплексов X–O2, сделанный на основании отнесения канала К5.
Рис. 15.
Схема резонансно-усиленной многофотонной ионизации РУМФИ [(1 + 1') + 1] синглетного кислорода с использованием двухцветной однолазерной схемы. Рисунок перепечатан из работы [68] с разрешения Elsevier.

Рис. 16.
Спектр РУМФИ [(1 + 1') + 1] молекулярного кислорода, образующегося при фотодиссоциации комплекса C5H8–O2: а – результаты эксперимента; б – результаты пересчета под использованную схему ионизации литературных спектров РУМФИ [2 + 1] кислорода в основном и синглетном состоянии, снятых ранее для других условий. Линии на рис. 16б, отмеченные символом ο, соответствуют сигналу несвязанного O2. Рисунок перепечатан из работы [68] с разрешения Elsevier.

Механизм образования синглетного кислорода. В качестве источника синглетного кислорода в работе [66] был предположен процесс с одновременным изменением электронного спина в молекулах-партнерах по комплексу
(XI)
$^{3}{{(}^{1}}{\text{X}}{\kern 1pt} - {{{\kern 1pt} }^{3}}{{{\text{O}}}_{2}}) + h{\nu } \to \,{{\,}^{3}}{\kern 1pt} {{(}^{3}}{\text{X}}{\kern 1pt} - {{{\kern 1pt} }^{1}}{{{\text{O}}}_{2}}) \to {{\,}^{3}}{\kern 1pt} {\text{X}} + {{\,}^{1}}{\kern 1pt} {{{\text{O}}}_{2}},$Рис. 17.
Схема электронно-колебательных состояний молекул в комплексе C2H4–O2, участвующих в процессе “двойной спин-флип” (XI). Рисунок адаптирован из работы [67] с разрешения журнала Phys. Chem. Chem. Phys.

Таблица 2.
Корреляция появления синглетного кислорода при фотодиссоциации комплексов X–O2 с оцениваемым положением максимума полосы перехода “двойной спин-флип”
| Комплекс X–O2 | Положение максимума полосы
“двойной спин-флип” ∆Еvert(3X ← 1X) + ∆Еvert(1O2 ← 3O2), эВ [67] |
Наличие сигнала ${{{\text{O}}}_{2}}\left( {{{a}^{1}}{{\Delta }_{g}}} \right)$ при фотодиссоциации комплекса (hν ≈ 5.5 эВ) [67] |
|---|---|---|
| C2H4–O2 | 5.5 | + |
| C3H6–O2 | 5.2 | + |
| C4H6–O2 | 4.2; 6.1а | + |
| C5H8–O2 | 4.2; 6.1а | + |
| C6H6–O2 | 5.1 | + |
| CH3I–O2 | 5.1б | + |
| CD3I–O2 | 5.1б | + |
| H2O–O2 | 8.0 | – |
| C6H12–O2 | 8.1 | – |
| Xe–O2 | 9.4 | – |
а Два значения соответствуют переходам X в триплетные состояния T1 и T2, соответственно. б Положение максимума пересчитано по сравнению с приведенным в работе [67] с учетом результатов работы [80], указывающих на то, что при ДСФ-переходе в комплексе CH3I–O2 имеет место переход в молекуле CH3I в триплетное состояние 3Q1 (∆Еvert(3X ← 1X) = 4.1 эВ [81]), а не 3Q0, как предполагалось в работе [67].
Как было отмечено выше, кинетическая энергия атомов кислорода, образующихся при фотодиссоциации синглетного кислорода (канал К5), очень близка к ожидаемой для фотодиссоциации колебательно холодного синглетного кислорода. Этот факт также имеет объяснение в рамках механизма “двойной спин-флип”. В комплексе X–O2 в основном электронном состоянии молекулы находятся довольно далеко друг от друга. Результаты ab initio расчета структуры комплексов X–O2 (X = C2H4 [82], CH3I, C6H12 и C3H4 [65]) показывают, что расстояние между центрами масс молекул X и O2 меняется в пределах 3.5–4 Å. Можно полагать, что на таком удалении структура уровней и вид волновых функций низколежащих возбужденных состояний молекулы X или O2 мало отличается от состояний свободной молекулы. Поэтому для переходов, локализованных на молекулах комплекса, можно использовать факторы ФК для свободных молекул. На рис. 17 показаны значения фактора ФК для переходов на разные колебательные уровни синглетного состояния ${{{\text{O}}}_{2}}({{a}^{{\,\,1}}}{{\Delta }_{g}}){\text{,}}$ рассчитанные для свободной молекулы O2 [83], которые показывают, что с вероятностью, близкой к 1, в результате вертикального возбуждения в синглетное состояние молекула O2 оказывается колебательно невозбужденной. Поэтому для комплексов X–O2 с любым X следует ожидать образования колебательно холодного синглетного кислорода ${{{\text{O}}}_{2}}({{a}^{{\,\,1}}}{{\Delta }_{g}})$ в результате ДСФ-перехода. Это и наблюдается в эксперименте. Приведенные результаты свидетельствуют в пользу ДСФ-механизма фотогенерации синглетного кислорода при возбуждении комплексов. В тоже время в работах, где регистрировалось образование синглетного кислорода при возбуждении в полосах супрамолекулярного поглощения в контактных комплексах кислорода и проводилось изучение новой фотохимии, его появление относили к возбуждению контактного комплекса X–O2 в состояние с переносом заряда [27, 31–34]. Поскольку контактные комплексы X–O2 в конденсированной среде и столкновительные комплексы в газовой фазе – это по сути Ван-дер-Ваальсовы комплексы молекул, то следует ожидать, что полученные выше выводы о ДСФ-механизме фотогенерации синглетного кислорода являются универсальными для всех этих комплексов. В тоже время среди исследованных комплексов есть и такие, для которых имеющиеся результаты позволяют сделать однозначный выбор между этими двумя механизмами. Так, для комплекса этилена, при возбуждении которого наблюдается образование синглетного кислорода, были рассчитаны структура комплекса и энергия состояния с переносом заряда [67]. Профили потенциальной энергии для состояний комплекса, участвующих в ДСФ- и ПЗ-переходах, показаны на рис. 18. Видно, что энергия фотона (hν ≈ 5.5 эВ) в эксперименте, в котором детектировалось образование синглетного кислорода, достаточна для ДСФ-перехода, но недостаточна для возбуждения ПЗ-состояния (∆Еvert$\left( {{\text{ПЗ}}} \right) = 7.7~\,\,{\text{эВ}}\,\,~\left[ {67} \right]$). В работе [84] Минаев с соавт. рассчитывали силы осцилляторов различных переходов в УФ-области спектра в столкновительных комплексах бензола с кислородом C6H6–O2, в том числе и перехода, который мы называем “двойной спин-флип”, при варьируемом межмолекулярном расстоянии. В случае, когда оно равно 3.6 Å, что cоответствует равновесному расстоянию в Ван-дер-Ваальсовом комплексе этилена с кислородом, такой переход становится доступным при энергии фотона, равной 5 эВ [84]. А его сила осциллятора составляет ~ 1% от самого интенсивного супрамолекулярного перехода с возбуждением кислорода в комплексе в состояние Герцберга III [84] (канал К1). Также авторы рассчитали энергию ПЗ-состояния при такой геометрии (7.2 эВ). При измерении карт скоростей атомов O(3P0), образующихся в результате фотодиссоциации Ван-дер-Ваальсовых комплексов C6H6–O2 под действием излучения с энергией фотона hν ≈ 5.5 эВ (λvac ≈ 226 нм), регистрировался канал К5, что указывает на образование синглетного кислорода ${{{\text{O}}}_{2}}({{a}^{{\,\,1}}}{{\Delta }_{g}})$ [67]. Таким образом, и в этом случае появление синглетного кислорода может быть объяснено ДСФ-процессом, но не переходом в ПЗ-состояние.
Рис. 18.
Сечение поверхности потенциальной энергии для конечного состояния перехода “двойной спин-флип” (ДСФ) и состояния с переносом заряда (ПЗ) комплекса C2H4–O2. Рисунок из работы [67] адаптирован с разрешения журнала Phys. Chem. Chem. Phys.

Здесь следует прокомментировать использование выражения (1)
(4)
${{E}_{{{\text{ПЗ}}}}} = ~{{I}_{{{\text{D}},{\text{vert}}}}} - {{A}_{{{\text{A}},{\text{vert}}}}} - \frac{{{{e}^{2}}}}{R}.$В работе [66] был выполнен двухлазерный эксперимент с измерением выхода синглетного кислорода в зависимости от длины волны излучения, возбуждающего комплекс изопрена с кислородом C5H8–O2 (рис. 19). Эта зависимость также отнесена к ДСФ-механизму возбуждения комплексов в процессе (XI), в котором молекула изопрена возбуждается во второй триплет ${}_{{}}^{3}{\text{X}} = {{{\text{C}}}_{5}}{{{\text{H}}}_{8}}\left( {{{{\text{T}}}_{2}}} \right)$. Приведенная в табл. 2 оценка максимума полосы ДСФ-перехода с возбуждением изопрена в триплет T2 соответствует энергии фотона 6.1 эВ (${{{\lambda }}_{{{\text{vac}}}}} \approx 203~$ нм). Для такого перехода сечение поглощение фотона и, соответственно, выход синглетного кислорода должны падать при увеличении длины волны, что находится в согласии с зависимостью на рис. 19.

Еще одно экспериментальное свидетельство в пользу ДСФ-механизма фотогенерации синглетного кислорода было получено в работе [80], где был исследован процесс фотодиссоциации комплекса CH3I–O2. Поскольку в ДСФ-процессе (XI) с образованием синглетного кислорода в состоянии ${{{\text{O}}}_{2}}({{a}^{1}}{{\Delta }_{g}})$ около 1 эВ идет на возбуждение кислорода, то, значит, триплет 3X должен образовываться с внутренней энергией, равной примерно hν – 1 эВ. Это предположение и проверяли в работе [80]. Была измерена кинетическая энергия атомов иода, появляющихся при фотодиссоциации комплексов CH3I–O2. Поскольку при фотовозбуждении молекулы CH3I избыток энергии над порогом диссоциации переходит в основном в кинетическую энергию фотофрагментов, то по измеренному ее значению можно восстановить величину внутренней энергии молекул CH3I, возбуждаемых в ДСФ-процессе. В результате возбуждения комплекса в условиях образования синглетного кислорода был зарегистрирован комплекс-специфический канал образования атомов иода I(2P3/2) с кинетической энергией, соответствующей диссоциации молекулы CH3I с энергией возбуждения меньшей, чем энергия фотона, на ~1 эВ. Этот результат также указывает на ДСФ-механизм образования синглетного кислорода.
Следует отметить, что поскольку спектральное положение перехода ДСФ отличается для различных X (табл. 2), то появляется возможность пространственно селективно (выбор по окружению) генерировать синглетный кислород в этом процессе, и, соответственно, запускать процесс фотоокисления.
Переход “двойной спин-флип” с образованием кислорода в синглетном состоянии ${{O}_{2}}\left( {{{b}^{1}}{\mathbf{\Sigma }}_{g}^{ + }} \right).~$ Канал К4. На рис. 6д, 7а и 7б в центре имиджа хорошо виден канал (К4) фотодиссоциации, соответствующий образованию атомов кислорода O(3PJ) с небольшой кинетической энергией в результате “параллельно” поляризованного перехода. По поляризации перехода, кинетической энергии атомов и характеру зависимости величины сигнала от квантового состояния (значения J) образующихся атомов O(3PJ) этот канал идентичен ранее наблюдавшемуся в работе [85] при фотодиссоциации молекулы кислорода во втором синглетном состоянии под действием излучения на этой же длине волны (~226 нм)
(XII)
${{{\text{O}}}_{2}}\left( {{{b}^{1}}{\Sigma }_{g}^{ + }} \right) + h{\nu } \to {\text{O}}{{{\text{(}}}^{3}}{{P}_{J}}{\text{)}} + {\text{O}}{{{\text{(}}}^{1}}D{\text{)}}{\text{.}}$Наличие канала К4 позволило сделать вывод об образовании синглетного кислорода при фотодиссоциации комплексов CH3I–O2 [64]. По аналогии с каналом К5 мы полагаем, что образование синглетного кислорода происходит в результате перехода (14) “двойной спин-флип”. Оценивая положение максимума полосы соответствующего перехода как сумму энергий вертикальных переходов ∆Еvert(3X ← 1X) + ∆Еvert(1O2 ← 3O2), для ДСФ-перехода $^{1}{\text{C}}{{{\text{H}}}_{3}}{\text{I}}{\kern 1pt} - {\kern 1pt} {{{\text{O}}}_{2}}\left( {{{b}^{1}}{\Sigma }_{g}^{ + }} \right)$ ← ${{~}^{1}}{\text{C}}{{{\text{H}}}_{3}}{\text{I}}{\kern 1pt} - {{{\kern 1pt} }^{3}}{{{\text{O}}}_{2}}\left( X \right)$ получаем значение 5.7 эВ, что очень близко к энергии возбуждающего фотона (hν ≈ 5.5 эВ) в обсуждаемых экспериментах.
Образование синглетного кислорода в результате возбуждения “возмущенного” состояния Герцберга. Не все случаи регистрации синглетного кислорода, объяснявшиеся ранее возбуждением комплекса X–O2 в ПЗ-состояние, определяются ДСФ-механизмом. Так, в работе [27] наблюдалось образование синглетного кислорода при возбуждении O2, растворенного в циклогексане, излучением с длиной волны 266 нм (hν = 4.66 эВ). На рис. 2б видно, что в этом растворе появляется полоса супрамолекулярного поглощения комплексов C6H12–O2. Но возбуждение Ван-дер-Ваальсовых комплексов C6H12–O2 в вышеуказанной полосе на 226 нм в молекулярном пучке не приводит к появлению синглетного кислорода (отсутствует канал К5 в картах скоростей образующихся атомов O(3PJ) [64]). И в соответствии с энергетическим критерием для ДСФ-механизма такой процесс невозможен при возбуждении излучением УФ-диапазона (табл. 2). При этом образование синглетного кислорода объясняется механизмом [67], который, как было показано ранее, реализуется в газовой фазе при возбуждении столкновительных комплексов кислорода X–O2 (X = O2, N2, C5H8) [86–88]. Этот механизм включает стадию усиленного поглощения в состояние Герцберга III кислорода в комплексах X–O2, что ведет к образованию возбужденных молекул ${{{\text{O}}}_{2}}(A{\kern 1pt} {{'}^{3}}{\kern 1pt} {{\Delta }_{u}}){\kern 1pt} :$
(XIII)
$\begin{gathered} {\text{X}}{\kern 1pt} - {\kern 1pt} {{{\text{O}}}_{2}}\left( {X{}_{{}}^{3}{\Sigma }_{g}^{ - }} \right)\xrightarrow{{h{\nu }}}{\text{X}}{\kern 1pt} - {\kern 1pt} {{{\text{O}}}_{2}}\left( {A{\kern 1pt} {\text{'}}{}_{{}}^{3}{{\Delta }_{u}}} \right) \to \\ \to {\text{X\;\;}} + {\text{\;}}{{{\text{O}}}_{2}}\left( {A{\kern 1pt} {\text{'}}{}_{{}}^{3}{{\Delta }_{u}}} \right). \\ \end{gathered} $Далее синглетный кислород образуется в последовательности процессов:
(XIV)
${{{\text{O}}}_{2}}\left( {A{\kern 1pt} {\text{'}}{}_{{}}^{3}{{\Delta }_{u}}} \right) \to {{{\text{O}}}_{2}}\left( {A{\kern 1pt} ',A,c} \right),$(XV)
$\begin{gathered} {{{\text{O}}}_{2}}\left( {A{\kern 1pt} ',A} \right) + {{{\text{O}}}_{2}}\left( {X{}_{{}}^{3}{\Sigma }_{g}^{ - }} \right) \to {{{\text{O}}}_{2}}(\{ a{}_{{}}^{1}{{\Delta }_{g}}\} , \\ \{ b{}_{{}}^{1}{\Sigma }_{g}^{ + }\} ) + {{{\text{O}}}_{2}}(\{ a{}_{{}}^{1}{{\Delta }_{g}}\} ,~\{ b{}_{{}}^{1}{\Sigma }_{g}^{ + }\} ). \\ \end{gathered} $Уравнение (XIV) соответствует быстрому столкновительному перераспределению заселенности между близко лежащими по энергии состояниями Герцберга $({{A}^{3}}\Sigma _{u}^{ + })$, $({{c}^{1}}\Sigma _{u}^{ - })$ и $(A{\kern 1pt} '{{~}^{3}}{{\Delta }_{u}}).$ Разрешенный по полному спину процесс (XV), детально обсуждаемый в работе [87], является аналогом триплет-триплетной аннигиляции, протекающей при генерации синглетного кислорода с помощью фотосенсибилизации [90, 91]. На рис. 20а показана спектральная зависимость квантового выхода синглетного кислорода по механизму (XIII)–(XV), в котором стадия (XIII) – это поглощение фотона столкновительным комплексом O2–O2. Спектр индуцированного столкновениями поглощения кислорода также показан на рис. 20б. В области длин волн короче ~242 нм (левее порога фотодиссоциации на рис. 20а) поглощение фотона сопровождается диссоциацией O2 на атомы, поэтому квантовый выход синглетного кислорода в этой области резко падает. При использовании излучения с длиной волны короче 242 нм генерируемые атомы кислорода рекомбинируют с молекулярным кислородом с образованием озона, фотодиссоциация которого также дает синглетный кислород [92]. Этот процесс хорошо известен и мы его в настоящем обзоре не рассматриваем.
Рис. 20.
а – Квантовый выход образования синглетного кислорода ${{{\text{O}}}_{2}}(a{}_{{}}^{1}{{\Delta }_{g}})$ при фотовозбуждении столкновительных комплексов O2–O2 в газовой фазе [87]. Штриховой стрелкой показано положение порога фотодиссоциации молекулы O2 на 41 269 см–1. б – Спектр поглощения, индуцированного столкновениями O2–O2 (полосы Вульфа). Стрелками указаны средние положения колебательных полос Вульфа v-0 [89]. Рисунок из работы [87] адаптирован с разрешения (2019) журнала J. Phys. Chem. A.

Характерной особенностью механизма (XIII)–(XV) является подобие спектра усиленного поглощения комплексов X–O2 спектру поглощения для переходов в состояния Герцберга в свободном O2. Граница этого поглощения с длинноволновой стороны находится вблизи 290 нм. Именно так выглядит спектр поглощения для кислорода в циклогексане (рис. 2б). Для всех исследованных Ван-дер-Ваальсовых комплексов X–O2, включая комплекс циклогексана C6H12–O2 [64], наблюдается канал (К1) возбуждения в состояние Герцберга III, соответствующий усиленному поглощению. Но в условиях экспериментов с молекулярным пучком из-за очень низкой концентрации частиц вторичные процессы (XIV) и (XV) невозможны. Поэтому в экспериментах по измерению карт скоростей отсутствует синглетный кислород, образующийся по механизму (XIII)–(XV). Но при возбуждении раствора кислорода в циклогексане молекулы кислорода, возбужденные в состояние Герцберга в процессах (XIII)–(XV), могут затем сталкиваться с растворенными молекулами кислорода в основном электронном состоянии и давать синглетный кислород в процессе (XV). Этот механизм должен универсально иметь место в газовой фазе (при не очень низком давлении кислорода, например в воздухе) и в растворах, содержащих кислород, при возбуждении в спектральной области полос Герцберга в O2 на длинах волн в диапазоне 242–290 нм. Реализация его может быть причиной и наблюдавшейся в работе [27] фотогенерации синглетного кислорода в алифатических алканах, содержащих растворенный кислород. Участием синглетного кислорода, образовавшегося по такому механизму, можно объяснить и разнообразные процессы фотоокисления, например, процессы фотодеградации нанокристаллических квантовых точек в гексане, протекающие в реальном технологическом процессе [93].
Полученные результаты позволяют нам сделать вывод, что образование синглетного кислорода в состояниях $^{1}{{{\text{O}}}_{2}}\left( {a,b} \right){\text{,}}$ зафиксированное при УФ-фотовозбуждении в новых полосах поглощения, появляющихся вследствие взаимодействия кислорода с молекулярным окружением, обусловлено двумя доминирующими механизмами. Один связан с возбуждением контактных или столкновительных комплексов X–O2 на переходах “двойной спин-флип”. Второй включает возбуждение молекул O2 в состояния Герцберга в результате поглощения в контактных или столкновительных комплексах X–O2, сопровождающееся образованием синглетного кислорода в результате триплет-триплетной аннигиляции при столкновении возбужденной и невозбужденной молекул O2. Вышеуказанные механизмы не связаны с возбуждением контактных или столкновительных комплексов X–O2 в состояние с переносом заряда, как считалось ранее. Это не значит, что возбуждение таких комплексов в ПЗ-состояние не приводит к генерации синглетного кислорода, но это требует изучения. Мы полагаем, что для возбуждения ПЗ-состояния исследованных комплексах X–O2 необходимо либо излучение ВУФ-диапазона, либо это возможно и с излучением УФ- и даже видимого диапазона при наличии полярного окружения, стабилизирующего ПЗ-состояние комплекса. Таким окружением для комплексов X–O2 могут служить полярный растворитель, ионы на поверхности и т.д.
Фотогенерация атомов водорода из комплексов X–O2. До этого шла речь преимущественно о каналах фотодиссоциации комплексов X–O2 с образованием атомарного или молекулярного кислорода. Наряду с этим наблюдались и комплекс-специфические процессы фотодиссоциации с появлением других продуктов. При фотодиссоциации комплексов алкенов с кислородом RH–O2 регистрировалось появление атомов водорода при возбуждении в спектральной области, где фотодиссоциация самих алкенов не обнаружена [94]. На рис. 21 представлены масс-спектры атомов водорода, которые демонстрируют наличие атома водорода в качестве фотофрагмента при УФ-фотодиссоциации Ван-дер-Ваальсовых комплексов алкенов с кислородом. В этой спектральной области присутствует крайне слабое поглощение алкенов на запрещенном синглет-триплетном переходе (3X ← 1X) гораздо ниже того уровня, который позволил бы наблюдать образование фрагментов в условиях эксперимента в работе [94]. Усиление поглощения на переходе 3X ← 1X в алкенах в присутствии растворенного кислорода впервые было обнаружено Эвансом [16]. Как показывают результаты работы [94], снятие запрета по полному спину на этот переход в комплексах с кислородом RH–O2, а также появление перехода “двойной спин-флип” определяют рост сечения поглощения в комплексах на порядки величины и появление атомов водорода в результате процессов:
(XVI)
$\begin{gathered} ^{3}{{(}^{1}}{\text{RH}}{\kern 1pt} --{{{\kern 1pt} }^{3}}{{{\text{O}}}_{2}}) + h\nu {{ \to }^{3}}{\kern 1pt} {{(}^{3}}{\text{RH}}{\kern 1pt} *{\kern 1pt} --{{{\kern 1pt} }^{3}}{{{\text{O}}}_{2}}) \to \\ \to \,\,{{{\kern 1pt} }^{3}}{\text{RH*}} + \,{{\,}^{3}}{{{\text{O}}}_{2}}, \\ \end{gathered} $(XVII)
$\begin{gathered} ^{3}{{(}^{1}}{\text{RH}}{\kern 1pt} --{{{\kern 1pt} }^{3}}{{{\text{O}}}_{2}}){\text{ }} + h\nu {{ \to }^{3}}{\kern 1pt} {{(}^{3}}{\text{RH*}}{\kern 1pt} --{{{\kern 1pt} }^{1}}{{{\text{O}}}_{2}}) \to \\ \to {\kern 1pt} {{\,}^{3}}{\kern 1pt} {\text{RH*}} + \,{{\,}^{1}}{\kern 1pt} {{{\text{O}}}_{2}}, \\ \end{gathered} $Рис. 21.
Масс-спектры атомов водорода, образующихся при фотодиссоциации Ван-дер-Ваальсовых комплексов RH–O2, где RH – этилен C2H4 (а), пропилен C3H6 (б), 2-бутен C4H8 (в) и 1,3-бутадиен C4H6 (а). Использовалось излучение с длиной волны 243.13 нм, которое соответствует линии резонансно-усиленной фотоионизации РУМФИ (2 + 1) атома H. Сигнал на m/e = 1 на нижних сканах (без O2) соответствует фоновому сигналу атомов H в камере масс-спектрометра. Рисунок перепечатан из работы [94] с разрешения Elsevier.

Следует ожидать, что в слабосвязанных комплексах кислорода X–O2 будут усиливаться и другие фотоиндуцируемые процессы, приводящие к доступным по энергии химическим превращениям в триплетном состоянии молекул X. Также передача энергии триплета 3X на кислород приводит к образованию синглетного кислорода.
4. ЗАКЛЮЧЕНИЕ
Полученные к настоящему времени результаты показывают, что взаимодействие кислорода со слабосвязанным окружением (столкновительные комплексы в газовой фазе, контактные комплексы в конденсированной среде) способно драматически влиять на скорость и механизм процессов фотоокисления. При этом молекулы окружения в результате могут оставаться неизменными, выполняя роль фотокатализатора, либо также претерпевать химические изменения. Большой прогресс в понимании природы влияния окружения достигнут при изучении фотохимии и фотофизики Ван-дер-Ваальсовых комплексов кислорода X–O2 в молекулярных пучках, в том числе с использованием техники измерения карт скоростей фотофрагментов.
Установлено, что влияние слабосвязанного партнера X по комплексу проявляется в драматическом увеличении сечения поглощения фотона кислородом в области полос Герцберга (200–290 нм). Усиление поглощения в комплексах X–O2 в этой области имеет место для любых X. От природы X зависит фактор усиления, который может достигать значений 106–107 раз. Усиленное поглощение в области континуума Герцберга (200–242 нм) приводит к возбуждению диссоциирующего электронного состояния Герцберга III. При этом реализуется и канал, в котором наряду с электронным возбуждением молекулы O2 происходит прямое колебательное возбуждение молекул X. За счет участия энергии внутренних степеней свободы молекул окружения X сдвигается в красную сторону спектральный порог фотодиссоциации молекул O2. Усиленное поглощение в дискретной области спектра в диапазоне 242–290 нм инициирует образование синглетного кислорода с квантовым выходом вплоть до значения φ ≈ 2, который является реакционным интермедиатом при фотоокислении.
В спектре слабосвязанных комплексов X–O2 появляются полосы супрамолекулярного поглощения, при котором одновременно возбуждаются обе молекулы комплекса с изменением электронного спина (“двойной спин-флип”). При этом также генерируется синглетное состояние кислорода, являющееся активным агентом фотоокисления. Возбуждаемое при этом триплетное состояние молекулы X также участвует в процессе фотоокисления, давая синглетный кислород (в результате передачи энергии) и другие реакционноспособные интермедиаты (атомы и радикалы). Следует отметить, что поскольку спектральное положение перехода “двойной спин-флип” отличается для различных X, то появляется возможность пространственно селективно (выбор по окружению) генерировать синглетный кислород, и, соответственно, запускать процесс фотоокисления.
Таким образом, полученные к настоящему времени результаты показывают, что учет супрамолекулярных процессов фотовозбуждения с участием кислорода и его слабосвязанного окружения является критически важным для оценки роли различных интермедиатов в процессах фотоокисления.
Список литературы
Parker D.H. // Acc. Chem. Res. 2000. V. 33. P. 563.
Wulf O.R. // Proc. Natl. Acad. Sci. (USA). 1928. V. 14. P. 609.
Finkelnburg W. // Z. Phys. 1934. V. 90. P. 1.
Blake A.J., McCoy D.G. // J. Quant. Spectrosc. Radiat. Transfer. 1987. V. 38. P. 113.
Shardanand // Phys. Rev. 1969. V. 186. P. 5.
Блок В.Р., Лебедев О.Л., Мехрякова Н.Г. // Докл. АН. 1979. Т. 249. С. 633.
Зеликина Г.Я., Берцев В.В., Киселева М.Б. // Опт. и спектр. 1994. Т. 77. № 4. С. 579.
Oshima Y., Okamoto Y., Koda S. // J. Phys. Chem. 1995. V. 99. P. 11830.
Зеликина Г.Я., Берцев В.В., Бурцев А.П., Кисе-лева М.Б. // Опт. и спектр. 1996. Т. 81. № 5. С. 751.
Зеликина Г.Я., Киселева М.Б., Бурцев А.П., Бер-цев В.В. // Опт. и спектр. 1998. Т. 85. № 4. С. 572.
Koda S., Sugimoto K. // J. Photochem. Photobiol. C: Photochem. Rev. 2003. V. 4. P. 215.
Goodman J., Brus L.E. // J. Chem. Phys. 1977. V. 67. P. 1482.
Бучаченко А.Л. Комплексы радикалов и молекулярного кислорода с органическими молекулами. Москва: Наука, 1984. 158 с.
Evans D.F. // J. Chem. Soc. 1953. P. 345.
Evans D.F. // J. Chem. Soc. 1957. P. 1351.
Evans D.F. // J. Chem. Soc. 1960. P. 1735.
Tsubomura H., Mulliken R.S. // J. Am. Chem. Soc. 1960. V. 82. P. 5966.
Hashimoto S., Akimoto H. // J. Phys. Chem. 1987. V. 91. № 6. P. 1347.
Hashimoto S., Akimoto H. // J. Phys. Chem. 1989. V. 93. № 2. P. 571.
Birks J.B., Pantos E., Hamilton T.D.S. // Chem. Phys. Lett. 1973. V. 20. P. 544.
Dijkgraaf C., Sitters R., Hoijtink G.J. // Mol. Phys. 1962. V. 5. №. 6. P. 643.
Dijkgraaf C., Hoijtink G.J. // Tetrahedron 1965. V. 19. Suppl. 2. P. 179.
Hoijtink G.J. // Acc. Chem. Res. 1969. V. 2. № 4. P. 114.
Minaev B.F., Kukueva V.V., Ågren H. // J. Chem. Soc. Faraday Trans. 1994. V. 90. P. 1479.
Minaev B.F., Mikkelsen K.V., Ågren H. // Chem. Phys. 1997. V. 220. P. 79.
Минаев Б.Ф. // Успехи химии. 2007. Т. 76. № 11. С. 1059.
Scurlock R.D., Ogilby P.R. // J. Phys. Chem. 1989. V. 93. P. 5493.
Slanger T.G., Jusinski L.E., Black G., Gadd G.E. // Science. 1988. V. 241. P. 945.
Андрухив В.И., Берцев В.В., Буланин М.О., Пастор А.А., Зеликина Г.Я., Сердобинцев П.Ю. // Оптика и спектр. 1994. Т. 76. № 1. С. 165.
Onodera K., Furusawa G., Kojima M., Tsuchiya M., Aihara S., Akaba R., Sakuragi H., Tokumaru K. // Tetrahedron. 1985. P. 2215.
Scurlock R.D., Ogilby P.R. // J. Am. Chem. Soc. 1988. V. 110. P. 640.
Kristiansen M., Scurlock R.D., Iu K.K., Ogilby P.R. // J. Phys. Chem. 1991. V. 95. P. 5190.
Jensen P.G., Arnbjerg J., Tolbod L.P., Toftegaard R., Ogilby P.R. // J. Phys. Chem. A. 2009. V. 113. P. 9965.
Дарманян А.П., Шарипов Д.Х. // Кинетика и катализ. 1990. Т. 31. С. 1486.
Singlet O2. Ed. Frimer A.A. CRC Press: Boca Raton, FL, 1985.
Singlet Oxygen: Applications in Biosciences and Nanosciences. Eds. Nonell S., Flors C. RSC Publishing, 2016.
Frei H. // Science. 2006. V. 313. P. 309.
Blatter F., Frei H. // J. Am. Chem. Soc. 1993. V. 115. P. 7501.
Sun H., Blatter F., Frei H. // J. Am. Chem. Soc. 1996. V. 118. P. 6873.
Blatter F., Sun H., Vasenkov S., Frei H. // Catal. Today. 1998. V. 41. P. 297.
Blatter F., Sun H., Frei H. // Catal. Lett. 1995. V. 35. P. 1.
Xu J., Mojet B.L., van Ommen J.G., Lefferts L. // J. Phys. Chem. B. 2004. V. 108. P. 15728.
Vanoppen D.L., De Vos D.E., Jacobs P.A. // Stud. Surf. Sci. Catal. 1997. V. 105. P. 1045.
Xiang Y., Larsen S.C., Grassian V.H. // J. Am. Chem. Soc.1999, V. 121, P. 5063.
Pidko E.A., van Santen R.A. // J. Phys. Chem. B 2006. V. 110. P. 2963.
Александров М.Л., Куснер Ю.С. // Газодинамические молекулярные, ионные и кластированные пучки. Л.: Наука, 1979. С. 270.
Milani P., Iannotta S. Cluster Beam Synthesis of Nanostructured Materials. Berlin: Springer, 1999.
DeBoer G., Young M.A. // J. Chem. Phys. 1997. V. 106. P. 5468.
DeBoer G., Prince A.P., Young M.A. // J. Chem. Phys. 2001. V. 115. P. 3112.
Parsons B.F., Chandler D.W. // J. Phys. Chem. A. 2003. V. 107. P. 10 544.
Giardini Guidoni A., Paladini A., Veneziani M., Naaman R., Di Palma T.M. // Appl. Surf. Sci. 2000. V. 154–155. P. 186.
Houston P.L., Chandler D.W. // J. Chem. Phys. 1987. V. 87. P. 1445.
Eppink A.T.J.B., Parker D.H. // Rev. Sci. Instrum. 1997. V. 68. P. 3477.
Imaging in molecular dynamics. Technology and applications. Ed. Whitaker B.J., Cambridge, University Press, 2003.
Ashfold M.N.R., Nahler N.H., Orr-Ewing A.J., Vieuxmaire O.P.J., Toomes R.L., Kitsopoulos T.N., Garcia I.A., Chestakov D.A., Wu S.M., Parker D.H. // Phys. Chem. Chem. Phys. 2006. V. 8. № 1. P. 26.
Chandler D.W., Houston P.L., Parker D.H. // J. Chem. Phys. 2017. V. 147. 013601.
Heck A.J.R., Chandler D.W. // Annu. Rev. Phys. Chem. 1995. V. 46. P. 335.
Eppink A.T.J.B., Parker D.H. // J. Chem. Phys. 1999. V. 110. P. 832.
Chestakov D., Parker D.H., Baklanov A.V. // J. Chem. Phys. 2005. V. 122. 084302.
Frederix P.W.J.M., Yang C.H., Groenenboom G.C., Parker D.H., K. Alnama, Western C.M., Orr-Ewing A.J. // J. Phys. Chem. A. 2009. V. 113. P. 14 995.
Zare R.N. // Mol. Photochem. 1972. V. 4. P. 1.
Buijsse B., van der Zande W.J., Eppink A.T.J.B., Parker D.H., Lewis B.R., Gibson S.T. // J. Chem. Phys. 1998. V. 108. P. 7229.
Chestakov D., Wu S.M., Wu G., Parker D.H., Eppink A.T.J.B., Kitsopoulos T.N. // J. Phys. Chem. A. 2004. V. 108. P. 8100.
Baklanov A.V., Bogdanchikov G.A., Vidma K.V., Chestakov D.A., Parker D.H. // J. Chem. Phys. 2007. V. 126. 124 316.
Vidma K.V., Bogdanchikov G.A., Baklanov A.V., Chestakov D.A., Parker D.H. // J. Chem. Phys. 2010. V. 133. 194 306.
Vidma K.V., Frederix P.W.J.M., Parker D.H., Baklanov A.V. // J. Chem. Phys. 2012. V. 137. 054 305.
Baklanov A.V., Bogomolov A.S., Pyryaeva A.P., Bogdanchikov G.A., Kochubei S.A., Farooq Z, Parker D.H. // Phys. Chem. Chem. Phys. 2015. V. 17. 28 565.
Bogomolov A.S., Dozmorov N.V., Kochubei S.A., Baklanov A.V. // Chem. Phys. Lett. 2018. V. 692. P. 271.
Minaev B.F., Mikkelsen K.V.,
 gren H. // Chem. Phys. 1997. V. 220. P. 79.
gren H. // Chem. Phys. 1997. V. 220. P. 79.
Mulliken R.S. // J. Am. Chem. Soc. 1952. V. 74. P. 811.
Aquilanti V., Ascenzi D., Cappelletti D., Castro M., Pirani F. // J. Chem. Phys. 1998. V. 109. P. 3898.
Ландау Л.Д., Лифшиц Е.М. Квантовая механика. Москва: Наука, 1974. 752 с.
Burns L.A., Vazquez-Mayagoitia A., Sumpter B.G., Sherrill C.D. // J. Chem. Phys. 2011. V. 134. 084 107.
Becucci M., Melandri S. // Chem. Rev. 2016. V. 116. P. 5014.
Chou P.T., Khan A.U. // Chem. Phys. Lett. 1984. V. 103. №. 4. P. 281.
Минаев Б.Ф. // Теорет. эксперим. химия. 1985. Т. 21. С. 594.
Свердлов Л.М., Ковнер М.А., Крайнов Е.П. Колебательные спектры многоатомных молекул. Москва: Наука, 1970. С. 560.
Farooq Z., Chestakov D.A., Yan B., Groenenboom G.C., van der Zande W.J., Parker D.H. // Phys. Chem. Chem. Phys. 2014. V. 16. P. 3305.
Schreiber M., Silva M.R., Sauer S.P.A., Thiel W. // J. Chem. Phys. 2008. V. 128. 134 110.
Bogomolov A.S., Kochubei S.A., Baklanov A.V. // Chem. Phys. Lett. 2016. V. 661. P. 53.
Gedanken A., Rowe M.D. // Chem. Phys. Lett. 1975. V. 34. P. 39.
Богданчиков Г.А., Бакланов А.В. // Журн. cтрукт. химии. 2015. Т. 56. № 5. С. 1041.
Nicholls R.W. // J. Res. Natl. Bur. Stand. Sect. A. 1965. V. 69A. P. 369.
Minaev B.F., Mikkelsen K.V., Ågren H. // Chem. Phys. 1997. V. 220. P. 79.
Eppink A.T.J.B., Parker D.H., Janssen M.H.M., Buijsse B., van der Zande W.J. // J. Chem. Phys. 1998. V. 108. P. 1305.
Trushina A.P., Goldort V.G., Kochubei S.A., Baklanov A.V. // Chem. Phys. Lett. 2010. V. 485. P. 11.
Trushina A.P., Goldort V.G., Kochubei S.A., Baklanov A.V. // J. Phys. Chem. A. 2012. V. 116. P. 6621.
Pyryaeva A.P., Goldort V.G., Kochubei S.A., Baklanov A.V. // Chem. Phys. Lett. 2014. V. 610. P. 8.
Fally S., Vandaele A.C., Carleer M., Hermans C., Jenouvrier A., Merienne M.F., Coquart B., Colin R. // J. Mol. Spectrosc. 2000. V. 204. P. 10.
Schmidt R. // Photochem. Photobiol. 2006. V. 82. P. 1161.
Krasnovsky A.A., Jr. // J. Phochem. Photobiol. A: Chem. 2008. V. 196. P. 210.
Окабе Х. Фотохимия малых молекул. Москва: Мир, 1981. 504 с.
Manner V.W., Koposov A.Y., Szymanski P., Klimov V.I., Sykora M. // ACS Nano. 2012. V. 6. P. 2371.
Baklanov A.V., Bogomolov A.S., Chikishev L.M., Bogdanchikov G.A., Kochubei S.A. // Chem. Phys. Lett. 2013. V. 558. P. 25.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ