

Химическая физика, 2021, T. 40, № 7, стр. 67-75
Моделирование адсорбции водорода на наночастице золота, нанесенной на подложку графита с различными дефектами
Н. В. Дохликова 1, *, А. К. Гатин 1, С. Ю. Сарвадий 1, С. А. Озерин 1, Е. И. Руденко 1, М. В. Гришин 1, Б. Р. Шуб 1
1 Федеральный исследовательский центр химической физики им. Н.Н. Семёнова
Российской академии наук
Москва, Россия
* E-mail: dohlikovanv@gmail.com
Поступила в редакцию 21.05.2020
После доработки 28.09.2020
Принята к публикации 20.10.2020
Аннотация
Методом квантово-химического моделирования системы наночастиц золота на подложке графита с различными дефектами показано понижение плотности состояний атомов золота при адсорбции водорода вблизи интерфейса. Дефекты подложки, такие как вакансии и обрывы плоскости графена, способствуют снижению плотности состояний при адсорбции атомов водорода.
ВВЕДЕНИЕ
Одни из наиболее перспективных типов структурированных на наноуровне покрытий образованы наночастицами, нанесенными на подложки различной природы. Варьируя элементный состав и размеры наночастиц, а также типы подложки, можно получать функциональные наноматериалы с гибкими и управляемыми свойствами [1]. Подложки могут оказывать достаточно разнообразное влияние на параметры наночастиц, их размеры и химический состав [2], оптические [3, 4] и химические свойства [5]. Вследствие этого изменяются поверхностное распределение наночастиц [6, 7], их зарядовое состояние [8] и характеристики химических реакций на поверхности [9]. Эти эффекты могут оказаться полезными как при создании элементов сложных систем, например, электроники с размером элементов <100 нм [10], так и при непосредственном использовании наноматериалов в качестве сенсоров [11, 12], катализаторов [13, 14] или сорбентов [15]. Взаимодействие наночастицы и подложки по сути можно упростить, выделив изменение атомной структуры наночастицы вследствие взаимодействия с подложкой и изменение ее заряда вследствие разности энергий Ферми свободной наночастицы и подложки. При этом необходимо помнить, что процессы трансформации атомной и электронной структур наночастицы при взаимодействии с подложкой влияют друг на друга. Указанное приближение до более явных дескрипторов вводится для интерпретации результатов исследования и прогнозирования свойств новых наносистем.
Если говорить о пространственном перераспределении электронов между наночастицей и подложкой, т.е. о приобретаемом заряде наночастицы, то знак заряда будет зависеть от энергий Ферми изолированных наночастицы и подложки [16]. Для наночастиц благородных и переходных металлов, в том числе и наночастиц золота, электронное строение удобно представлять в рамках модели центра d-зоны [17]. Соответственно, если наночастица приобретает отрицательный заряд, то “центр тяжести” заполненных состояний в случае d-орбиталей атомов Au сдвигается вверх по энергии в сравнении с нейтральным металлическим кластером. Если же наночастица заряжена положительно, то ситуация обратная. Однако взаимодействие с подложкой также может быть причиной достаточно существенных изменений атомного строения наночастицы, которые сопровождаются эволюцией электронной структуры. В целом эти факторы взаимосвязаны и влияют на физико-химические свойства наночастицы совместно.
На графитовой подложке можно ожидать присутствие различных дефектов, как единичных, так и многоатомных, которые также оказывают влияние на атомную и электронную подсистему нанесенной наночастицы [18]. Исследование адсорбционных свойств такой системы на примере атома H актуально не только в прикладном, но и в фундаментальном смысле, поскольку подложка графита представляет собой поверхность с хорошо определенной атомной структурой, а атом H является типичным модельным объектом при исследовании адсорбции [19].
Однако наносистемы на основе наночастиц Au на графитовой подложке также исследованы и в более сложных реакциях. Например, в работе [20] рассмотрен электрокатализ Н2О2 с помощью композитной пленки на основе восстановленного оксида графена, наночастиц Au и полиоксометаллата. Полученный нанокомпозит с наночастицами Au обладал большей электрокаталитической активностью, чем нанокомпозит с одним полиоксометалатом. Авторы связывают такое улучшение каталитических свойств с синергетическим эффектом подложки и наночастиц. Похожий нанокомпозит на основе анионных наночастиц Au и кластеров полиоксиметалата на восстановленном оксиде графена исследован как биосенсор мочевой кислоты в работе [21]. Анализ показал, что описанная выше система обладает приемлемыми аналитическими характеристиками, стабильностью и селективностью благодаря высокой диффузии и передаче заряда. В работе [22] созданы и протестированы новые, сверхчувствительные биосенсоры для обнаружения ДНК, представляющие собой графен с нанесенными на него наночастицами Au. Установлено, что с помощью синтезированных наносистем можно достичь чувствительности этих сенсоров до 0.057 фмоль. Можно заключить, что добавление наночастиц Au в состав нанокомпозита повышает его проводящие и реакционные свойства, что актуально для практического применения.
Кроме того, структура примесей золота в составе биметаллических кластеров также широко изучена. Например, подробное исследование наночастиц Au/Pd, нанесенных на графен, показало [23], что кластеры каталитических биметаллических наночастиц представляют собой ядра Au, заключенные в оболочку Pd. Несмотря на то, что катализаторы гидрирования на основе палладия давно известны, эти исследования актуальны как для более эффективного синтеза промышленных катализаторов, так и для лучшего понимания микроскопических механизмов элементарных реакций.
Исследование взаимодействия графена с золотом на уровне одного внедренного атома Au проведено в работе [24] на примере каталитической реакции гидрохлорирования ацетилена. Моделирование механизма реакции методом теории функционала плотности (DFT) показало, что добавление атома азота может снизить энергию активации. В работе [25] исследовано взаимодействие наночастицы золота с графитовой подложкой на примере наносистем кластера золота Au12 и полициклических ароматических углеводородов (ПАУ) C6H6, C24H12, C54H16, C96H18. Результаты исследований показали, что характер взаимодействия в системе Au–ПАУ на 45% дисперсионный, на 35% электростатический и на 20% ковалентный. Однако авторы этих работ отмечают, что влияние ПАУ на кластер зависит также от размера самого ПАУ.
В наших предыдущих работах с помощью похожей кластерной модели уже исследовалось влияние графитовой подложки на адсорбцию атомов Н на наночастицах, смоделированных кластерами Au13. В работе [26] для имитации воздействия подложки графита использована наночешуйка графена, а в работе [27] с помощью двух наночешуек графена разного размера, расположенных параллельно друг другу, смоделирован обрыв графитовой плоскости. В настоящей работе проведено исследование влияния графитовой подложки, в том числе и с дефектами, на адсорбционные свойства нанесенной наночастицы Au в периодических условиях. Рассмотренные поверхностные дефекты включали в себя одноатомную вакансиию, поворот связи С–С на 90° или топологический дефект 5-7, обрыв плоскости графена типа “кресло” и “зигзаг”. Также было исследовано влияние подвижности атомов Au на взаимодействие c атомом H, для чего расчеты проводились при фиксированном и незакрепленном положении атомов Au.
МЕТОДИКА ИССЛЕДОВАНИЙ
Моделирование наносистемы, имитирующей наночастицу Au, нанесенную на графитовую подложку, проводилось с использованием метода DFT. Модель подложки представляла собой две графеновых плоскости, содержащих по шесть краевых атомов с каждой стороны. Расстояние между плоскостями после оптимизации атомной структуры с варьированием параметров ячейки равно 4.34 Å, объем расчетной ячейки – 24 ⋅ 103 Å3.
В данном исследовании рассматривались следующие дефекты подложки: вакансия атома углерода, дефект 5-7, обрывы плоскости графена типа “кресло” и “зигзаг”. Во всех случаях атомная структура верхней плоскости графена была дополнительно оптимизирована без варьирования параметров ячейки для устранения напряжений в структуре при соблюдении единообразия размеров расчетной ячейки.
В качестве исходной модели наночастицы Au использовался 13-атомный икосаэдрический изомер кластера Au. Атомная структура, т.е. взаимное расположение атомов кластера друг относительно друга, рассчитывалась при фиксированном положении всех атомов подложки. Поскольку данная работа посвящена исследованию вопросов, касающихся выявления изменений электронной структуры адсорбционного комплекса под влиянием подложки, полученная наносистема Au–графит, соответствующая локальному энергетическому минимуму, удовлетворяет поставленным требованиям. Ожидаемо, что кластеры в наносистемах с обрывом плоскости графита подвергаются большему воздействию, из-за чего их атомная структура испытывает значительную трансформацию, особенно в системе с обрывом плоскости типа “зигзаг” (табл. 1). Изменения в системах с графитом без дефектов, с вакансией и дефектом 5-7 намного менее выраженные. Полученные результаты моделирования полностью соответствуют экспериментальным данным из работ [28–32]. Из этих данных следует, что металлические и металлоксидные наночастицы концентрируются вблизи краев графеновых листов, образующих графитовую подложку.
Таблица 1.
Величины энергий связи 13-атомных кластеров Au с подложками графита без дефекта (1), с вакансией (2), с дефектом Стоуна–Уэльса (3), обрыв плоскости графена типа “кресло” (4), обрыв плоскости графена типа “зигзаг” (5)
| Подложка | Энергия связи, эВ |
|---|---|
| 1 | –0.16 |
| 2 | –0.42 |
| 3 | –0.18 |
| 4 | –1.60 |
| 5 | –1.76 |
Анализ заселенности по Малликену и построение распределений электронной плотности наносистемы Au–графит показали накопление небольшого количества отрицательного заряда в кластере Au. Взаимное влияние кластеров друг на друга исследовано с помощью тестовых расчетов с варьированием размеров ячейки, т.е. расстояния между кластерами. Эти расчеты показали, что взаимное влияние кластеров в условиях наших расчетов пренебрежимо мало.
Моделирование атомной и электронной структуры проводилось с использованием свободных программных пакетов Quantum Espresso (QE) [33] и OpenMX (OMX) [34]. Для расчета электронной структуры использовано приближение локальной плотности (LDA) в ультрамягком псевдопотенциале, взятом из стандарной библиотеки псевдопотенциалов программного пакета QE. Данные условия расчета были выбраны на основании тестовых вычислений, которые показали лучшую сходимость по электронной подсистеме в исследуемой наносистеме Au–графит. Радиусы обрезания псевдопотенциалов атомов наносистемы выбраны исходя из рекомендованных в документации файлов псевдопотенциалов. Наборы атомноцентрированных базисных функций взяты из документации к программному пакету OMX.
Места адсорбции атомов водорода в системе (ниже для них будет использован термин “сайт”) определялись как при фиксированном, так и при незакрепленном положении атомов Au, что было необходимо для исследования влияния подвижности атомов Au на энергию связи с водородом. Для каждой системы проводилось моделирование различных сайтов: нескольких в окрестности границы кластера Au и графита (далее – вблизи интерфейса) и одного на вершине кластера, вдали от интерфейса. Для кластера Au на бездефектной подложке графита, подложке графита с атомной вакансией и подложке графита с дефектом 5-7 исследованы четыре сайта адсорбции: s1 – на вершине кластера и s2–s4 – вблизи интерфейса, вокруг кластера.
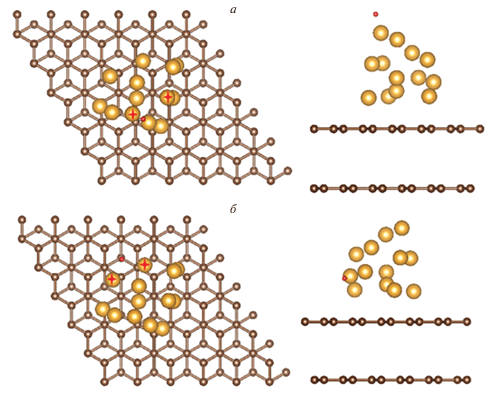
Для кластера Au, расположенного на краю оборыва плоскости графена типа “кресло” и “зигзаг”, исследованы три сайта: s1–s3. В системе с обрывом плоскости типа “кресло” это сайты на вершине кластера s1, вблизи интерфейса над плоскостью графита s2 и вблизи интерфейса под плоскостью графита s3. В системе с обрывом типа “зигзаг” это сайты на вершине кластера s1 и два различных вблизи интерфейса – s2, s3. Примеры типичных сайтов приведены на рис. 1.
Рис. 1.
Примеры выбранных сайтов адсорбции атома Н на плоскости графита: а – на вершине кластера на подложке без дефектов, б – вблизи интерфейса на подложке без дефектов.
Расчет изменения спроектированной плотности состояний отдельных атомов вблизи сайта адсорбции атома H является важной частью исследования, поскольку позволяет провести сопоставление результатов экспериментов с использованием методов сканирующей туннельной микроскопии (СТМ) и спектроскопии (СТС) и моделирования. Для получения более подробной информации отдельно изучено поведение s- и d-орбиталей атомов золота. В предыдущих работах мы показали, что именно состояния атомов в окрестности уровня Ферми вносят основной вклад в изменение туннельного тока в СТС-спектроскопии [26, 27]. При небольших напряжениях смещения U туннельный ток I будет определяться только плотностью состояний образца:
(1)
$I(U) = {\text{const}} \cdot M{{\rho }_{{tip}}}(0)\int\limits_{ - eU}^0 {{{\rho }_{{sample}}}(\varepsilon )d\varepsilon } .$Здесь M – усредненное значение матричного элемента перекрывания волновых функций зонда и атомов поверхности, ρtip(0) – аппроксимированное значение плотности состояний зонда вблизи уровня Ферми, ρsample(ε) – локальная плотность состояний образца, ε – собственные значения энергии.
Заметим, что влияние адсорбции на распределение плотности поверхностных состояний хорошо известно [16]. Именно этот эффект уменьшения плотности состояний в окрестности уровня Ферми, наблюдающийся в СТМ/СТС-экспериментах с системой Au–H, и позволяет детектировать акты взаимодействия (диссоциативной адсорбции) H с наночастицами Au.
КВАНТОВО-ХИМИЧЕСКОЕ МОДЕЛИРОВАНИЕ
Определено влияние подвижности атомов золота на величину энергии связи Au–H. Для системы с подложкой без дефекта это влияние наиболее заметно для сайта s1 (табл. 2). Моделирование адсорбции атомов H на подложках с единичными дефектами показало, что величины энергий связи вблизи интерфейса и на вершине различаются незначительно (табл. 3, 4), но для всех систем большую роль играет подвижность атомов Au. Можно отметить, что для системы с подложкой, содержащей вакансию атома углерода, энергии связи для сайтов вблизи интерфейса и на вершине в случае подвижных атомов Au практически не различаются, а наибольшее влияние подвижность атомов Au оказывает на энергию связи сайта s3 вблизи интерфейса (табл. 3). Для системы с подложкой с дефектом 5-7 подвижность атомов Au влияет на энергию связи более равномерно (табл. 4). Как видно, величина последней в данных условиях сильно зависит от локального окружения.
Таблица 2.
Энергия и длина связи для сайтов адсорбции атома H на кластере Au на бездефектной подложке при фиксированном положении атомов Au, Eсв и Rсв, и при нефиксированном положении атомов Au, $E_{{св}}^{*}$ и $R_{{св}}^{*}$
| Сайт | Eсв, эВ | Rсв, Å | $E_{{{\text{св}}}}^{*}$, эВ | $R_{{{\text{св}}}}^{*}$, Å | Eсв – $E_{{{\text{св}}}}^{*}$, эВ |
|---|---|---|---|---|---|
| s1 | –3.34 | 1.62 | –3.94 | 1.80; 1.81 | 0.60 |
| s2 | –3.49 | 1.76; 1.78 | –3.67 | 1.78; 1.77 | 0.18 |
| s3 | –3.35 | 1.88; 1.71 | –3.55 | 1.72; 1.87 | 0.20 |
| s4 | –3.29 | 1.73; 1.86 | –3.46 | 1.84; 1.72 | 0.17 |
Таблица 3.
Энергия и длина связи для сайтов адсорбции атома H на кластере Au на подложке с единичной вакансией при фиксированном положении атомов Au, Eсв и Rсв, и при нефиксированном положении атомов Au, $E_{{св}}^{*}$ и $R_{{св}}^{*}$
| Сайт | Eсв, эВ | Rсв, Å | $E_{{{\text{св}}}}^{*}$, эВ | $R_{{{\text{св}}}}^{*}$, Å | Eсв – $E_{{{\text{св}}}}^{*}$, эВ |
|---|---|---|---|---|---|
| s1 | –3.41 | 1.60 | –3.75 | 1.81; 1.77 | 0.34 |
| s2 | –3.27 | 1.84; 1.74 | –3.40 | 1.90; 1.73 | 0.13 |
| s3 | –2.98 | 1.81; 1.74 | –3.73 | 1.80; 1.70 | 0.75 |
| s4 | –3.64 | 1.72; 1.80 | –3.76 | 1.72; 1.81 | 0.12 |
Таблица 4.
Энергия и длина связи для сайтов адсорбции атома H на кластере Au на подложке графита с дефектом Стоуна–Уэльса при фиксированном положении атомов Au, Eсв и Rсв, и при нефиксированном положении атомов Au, $E_{{св}}^{*}$ и $R_{{св}}^{*}$
| Сайт | Eсв, эВ | Rсв, Å | $E_{{{\text{св}}}}^{*}$, эВ | $R_{{{\text{св}}}}^{*}$, Å | Eсв – $E_{{{\text{св}}}}^{*}$, эВ |
|---|---|---|---|---|---|
| s1 | –3.45 | 1.62 | –3.84 | 1.80; 1.79 | –0.39 |
| s2 | –3.20 | 1.75; 1.83 | –3.58 | 1.80; 1.76 | –0.38 |
| s3 | –3.12 | 1.62; 2.64 | –3.52 | 1.94; 1.68 | –0.40 |
| s4 | –3.38 | 1.79; 1.74 | –3.79 | 1.77; 1.77 | –0.41 |
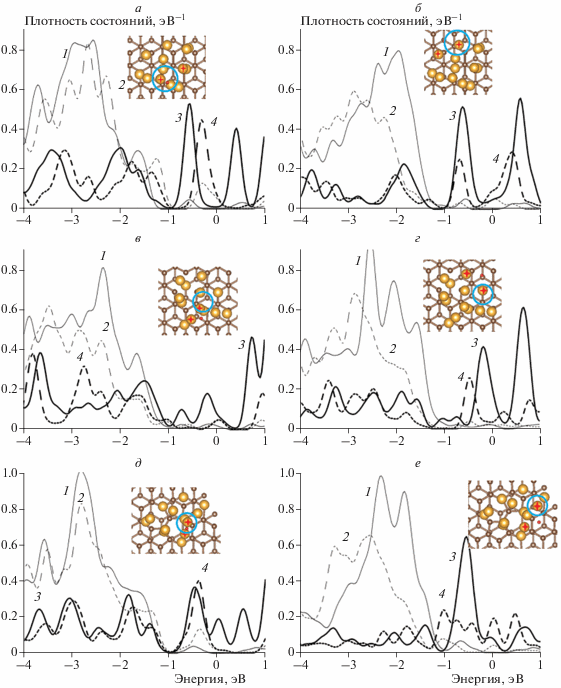
На рис. 2 приведены рассчитанные распределения спроектированных плотностей состояний s- и d-орбиталей атомов Au различных сайтов адсорбции атома H в трех системах: с бездефектным графитом, графитом с вакансией и графитом с дефектом 5-7. Для краткости представлены два типа сайтов: один на вершине и один вблизи интерфейса для каждой подложки соответственно. Расчет показал, что плотность состояний во всех описанных выше системах вблизи уровня Ферми образована s-орбиталями, а “зона” d-орбиталей расположена ниже по энергии, как и в свободных 13-атомных кластерах [35] (рис. 2). Для сайтов в окрестности интерфейса во всех системах характерен сдвиг вниз по энергии плотности состояний d-орбиталей и снижение плотности состояний s-орбиталей вблизи уровня Ферми. Кроме того, наблюдается усиление этой тенденции при приближении к подложке в наносистемах с бездефектным графитом и графитом с дефектом 5-7. Для сайта s1 в системе с бездефектной подложкой происходит сдвиг плотности состояний вверх по энергии. В системе с дефектом 5-7 изменение плотности состояний для сайта s1 незначительно. Однако для сайта s1 в системе с подложкой с вакансией плотность состояний изменяется так же, как и для сайтов вблизи интерфейса. В целом это не противоречит экспериментальным данным, согласно которым уменьшение туннельного тока наблюдалось именно вблизи интерфейса [26, 27]. Также можно отметить, что, несмотря на различия атомных структур кластеров Au на различных подложках, обнаруживается одинаковое влияние подложки на изменение электронной структуры кластера при адсорбции H: близость подложки усиливает снижение плотности состояний, а наличие дефектов увеличивает этот эффект.
Рис. 2.
Спроектированные плотности состояний: 1 – d-орбиталей атомов Au до адсорбции H, 2 – d-орбиталей атомов Au после адсорбции H, 3 – s-орбиталей атомов Au до адсорбции H, 4 – s-орбиталей атомов Au после адсорбции H. Cайты на бездефектном графите: а – на вершине, б – вблизи интерфейса, на графите с вакансией; в – на вершине; г – вблизи интерфейса, на графите с дефектом 5-7; д – на вершине; е – вблизи интерфейса. Атомы Au, ближайшие к атому H, помечены крестиками. Атомы Au, для которых проводился расчет плотностей состояний, отмечены кружками.
Моделирование адсорбции H на подложках с обрывами плоскостей графена показало, что в данных системах энергии связи Au–H для сайтов вблизи интерфейса и на вершине различаются сильно (табл. 5, 6). Поскольку энергия связи на интерфейсе по модулю меньше энергии связи на вершине примерно на 1 эВ, можно предположить, что атом H образует связь с атомами C и Au. Также длины связи указывают на то, что при адсорбции вблизи интерфейса атом H связывается “мостиком” через атомы C и Au. Исследование влияния подвижности атомов Au на адсорбцию H выявило сильное влияние этого фактора для отдельных сайтов s2 вблизи интерфейса для системы с обрывом плоскости типа “кресло” и s3 для системы с обрывом плоскости типа “зигзаг”. Это может указывать на сильную нестабильность адсорбции H на сайтах вблизи интерфейса, что не противоречит экспериментальным данным по длительной релаксации вольт-амперных характеристик.
Таблица 5.
Энергия и длина связи для сайтов адсорбции атома H на кластере Au на подложке с обрывом плоскости графена типа “кресло” при фиксированном положении атомов Au, Eсв и Rсв, и при нефиксированном положении атомов Au, $E_{{св}}^{*}$ и $R_{{св}}^{*}$
| Сайт | Eсв, эВ | Rсв, Å | $E_{{{\text{св}}}}^{*}$, эВ | $R_{{{\text{св}}}}^{*}$, Å | Eсв – $E_{{{\text{св}}}}^{*}$, эВ |
|---|---|---|---|---|---|
| s1 | –3.50 | 1.58 | –3.70 | 1.72; 1.81 | –0.20 |
| s2 | –2.24 | 1.25; 2.05; 1.82 | –3.57 | 2.00; 1.59; 1.11 | –1.33 |
| s3 | –2.35 | 1.30; 1.72; 2.38 | –3.26 | 1.14; 2.13 | –0.91 |
Таблица 6.
Энергия и длина связи для сайтов адсорбции атома H на кластере Au на подложке с обрывом плоскости графена типа “зигзаг” при фиксированном положении атомов Au, Eсв и Rсв, и при нефиксированном положении атомов Au, $E_{{св}}^{*}$ и $R_{{св}}^{*}$
| Сайт | Eсв, эВ | Rсв, Å | $E_{{{\text{св}}}}^{*}$, эВ | $R_{{{\text{св}}}}^{*}$, Å | Eсв – $E_{{{\text{св}}}}^{*}$, эВ |
|---|---|---|---|---|---|
| s1 | –4.04 | 1.75; 1.76 | –4.16 | 1.75; 1.76 | –0.12 |
| s2 | –3.57 | 1.17; 1.83; 2.82 | –4.18 | 1.12; 2.77; 2.09 | –0.61 |
| s3 | –3.47 | 1.16; 1.86; 2.28 | –4.79 | 1.11; 2.46; 2.33 | –1.32 |
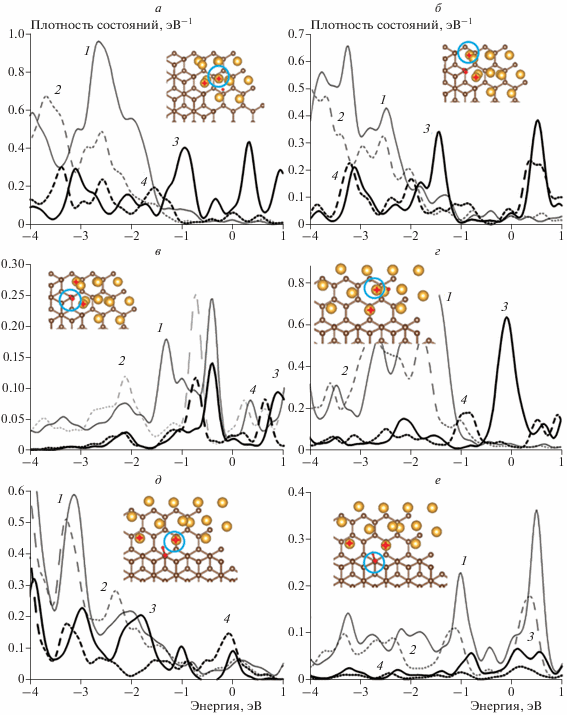
На рис. 3 приведены рассчитанные распределения спроектированных плотностей состояний s- и d-орбиталей атомов Au и s- и p-орбиталей атомов C различных сайтов адсорбции атома H в двух системах с обрывами плоскостей графена типа “кресло” или типа “зигзаг”. Для краткости представлены два типа сайтов: один на вершине и один вблизи интерфейса для каждой подложки соответственно. Расчет показал, что распределение плотностей состояний атомов Au в обеих системах изменяется намного сильнее, очевидно, благодаря большему взаимодействию с граничными атомами C. Распределение плотности состояний d-орбиталей сдвигается вниз по энергии под воздействием подложки. Разделение d- и s‑орбиталей уже не столь явное (рис. 3). Однако, несмотря на это, распределение плотности состояний атомов Au изменяется характерным для свободного кластера образом: сдвиг вниз по энергии плотностей состояний d-орбиталей и снижение плотности состояний s-орбиталей вблизи уровня Ферми как для сайтов на вершинах, так и для сайтов вблизи интерфейса. Плотности состояний p- и s-орбиталей атома C также сдвигаются вниз, что указывает на двухцентровую связь атома H.
Рис. 3.
Спроектированные плотности состояний: 1 – d-орбиталей атомов Au и p-орбиталей атомов С до адсорбции H, 2 – d-орбиталей атомов Au и p-орбиталей атомов С после адсорбции H, 3 – s-орбиталей атомов Au и С до адсорбции H, 4 – s-орбиталей атомов Au и С после адсорбции H. Сайты на обрыве плоскости графена типа “кресло”: а – на вершине; б, в – вблизи интерфейса, на обрыве плоскости графена типа “зигзаг”; г – на вершине; д, е – вблизи интерфейса. Атомы Au и С, ближайшие к атому H, помечены крестиками. Атомы Au и С, для которых проводился расчет плотностей состояний, отмечены кружками.
ВЫВОДЫ
1. Во всех наносистемах наблюдалось снижение плотности состояний атомов Au на сайтах адсорбции атома H вблизи интерфейса.
2. На величину энергии связи большое влияние оказывает подвижность атомов Au.
3. Дефекты подложки, такие как вакансии и обрывы плоскости графена, способствуют снижению плотности состояний при адсорбции атомов H, даже в случае сайтов вдали от интерфейса.
Все расчеты выполнены с использованием вычислительных ресурсов Межведомственного Суперкомпьютерного Центра РАН (МСЦ РАН).
Работа выполнена в рамках госзадания (регистрационный номер АААА-А20-120013190076-0), а также при финансовой поддержке Российским фондом фундаментальных исследований (грант № 18-03-00060).
Список литературы
Lu F., Astruc D. // Coord. Chem. Rev. 2020. V. 408. P. 31.
Molodtsova O.V., Aristova I.M., Potorochin D.V. et al. // Appl. Surf. Sci. 2020. V. 506. P. 8.
Nevruzoglu V., Altuntas D.B., Tomakin M. // Appl. Phys. A. 2020. V. 126. № 4. P. 9.
Журавлева Т.С., Иванова О.П., Криничная Е.П. и др. // Хим. физика. 2011. Т. 30. № 8. С. 75.
Magnin Y., Villermaux E., Amara H. et al. // Carbon. 2020. V. 159. P. 504.
Jiang R.M., Li W.X., Li Y. et al. // Appl. Phys. Lett. 2020. V. 116. № 10. P. 4.
Рыбальтовский А.О., Аракчеев В.Г., Минаев Н.В. и др. // Сверхкритич. флюиды: теория и практика. 2019. Т. 14. № 1. С. 47.
He F., Wang Z.X., Wei S.Q. et al. // Appl. Surf. Sci. 2020. V. 506. P. 7.
Lee M.Y., Ha H., Cho K.H. et al. // ACS Catal. 2020. V. 10. № 2. P. 1237.
Choi H., Nguyen P.T., Tran P.V. et al. // Appl. Surf. Sci. 2020. V. 510. P. 6.
Liu R.Z., Xu K., Zhang Y. // Instrum. Sci. Technol. 2020. V. 48. № 4. P. 459.
Желтова А.В., Смолянский А.С., Бирюков Ю.Г. и др. // Хим. физика. 2018. Т. 37. № 10. С. 72.
Bhaduri B., Polubesova V. // Mat. Lett. 2020. V. 267. P. 4.
Ракитин М.Ю., Долуда В.Ю., Тянина А.А. и др. // Сверхкритич. флюиды: теория и практика. 2016. Т. 11. № 3. С. 10.
Ильясова Р.Р., Гайнетдинова Ю.М., Массалимов И.А. и др. // Хим. физика. 2017. Т. 36. № 8. С. 90.
Николаевич Н.Н. Технология конструкционных материалов. Анализ поверхности методами атомной физики. Уч. пос. 1-е изд. М.: Изд-во “Юрайт”, 2018.
Hammer B., Norskov J.K. // Surf. Sci. 1996. V. 359. № 1–3. P. 306.
Backes C., Abdelkader A.M., Alonso C. et al. // 2D Mater. 2020. V. 7. № 2. P. 282.
Kulkarni A., Siahrostami S., Patel A. et al. // Chem. Rev. 2018. V. 118. № 5. P. 2302.
Zhang X.X., Bao Y.Y., Bai Y.F. et al. // Electrochim. Acta. 2019. V. 300. P. 380.
Bai Z.Y., Zhou P.L., Xu H.B. et al. // Sens. Actuators, B. 2017. V. 243. P. 361.
Wang W., Bao T., Zeng X. et al. // Biosens. Bioelectron. 2017. V. 91. P. 183.
Meduri K., Stauffer C., Johnson G.O. et al. // Microsc. Microanal. 2019. V. 25. № 1. P. 80.
Gong W.Q., Zhao F., Kang L.H. // Comput. Theor. Chem. 2018. V. 1130. P. 83.
Munoz-Castro A., Gomez T., Carey D.M. et al. // J. Phys. Chem. P. 2016. V. 120. № 13. P. 7358.
Дохликова Н.В., Колченко Н.Н., Гришин М.В. и др. // Рос. нанотехнол. 2016. Т. 11. № 11–12. С. 54.
Gatin A.K., Grishin M.V., Dokhlikova N.V. et al. // Crystals. 2019. V. 9. № 7. P. 14.
Гатин А.К., Гришин М.В., Дохликова Н.В. и др. // Докл. АН. 2016. Т. 470. № 1. С. 60.
Sarvadiy S.Yu., Gatin A.K., Grishin M.V. et al. // Gold Bull. 2019. V. 52. № 2. P. 61.
Гришин М.В., Гатин А.К., Дохликова Н.В. и др. // Кинетика и катализ. 2015. Т. 56. № 4. P. 539
Гришин М.В., Гатин А.К., Сарвадий С.Ю. и др. // Рос. нанотехнол. 2017. Т. 12. № 11–12. С. 15.
Гатин А.А., Гришин М.В., Дохликова С.В. и др. // Рос. нанотехнол. 2018. Т. 13. № 9–10. С. 3.
Giannozzi P., Andreussi O., Brumme T. et al. // J. Phys.: Condens. Matter. 2017. V. 29. № 46. P. 30.
Ozaki T., Kino H. // Phys. Rev. B. 2004. V. 69. № 19. P. 19.
Дохликова Н.В., Колченко Н.Н., Гришин М.В. и др. // Рос. нанотехнол. 2016. Т. 11. № 1–2. С. 17.
Дополнительные материалы отсутствуют.
Инструменты
Химическая физика