

Журнал аналитической химии, 2019, T. 74, № 2, стр. 97-102
ИСПОЛЬЗОВАНИЕ ОБРАЩЕННО-ФАЗОВОЙ ВЭЖХ ДЛЯ КАЧЕСТВЕННОГО И КОЛИЧЕСТВЕННОГО КОНТРОЛЯ ПОЛУЧЕНИЯ N-ОКТАДЕЦИЛ-1,3-ДИАМИНОПРОПАНА
М. А. Солдатов 1, *, В. М. Носова 1, Е. А. Монин 1, П. А. Стороженко 1
1 Государственный научно-исследовательский институт химии и технологии элементоорганических соединений (ГНИИХТЭОС)
105118 Москва, шоссе Энтузиастов, 38, Россия
* E-mail: soldat89.89@gmail.com
Поступила в редакцию 23.11.2017
После доработки 11.05.2018
Принята к публикации 11.05.2018
Аннотация
Методом обращенно-фазовой ВЭЖХ разработан способ качественного и количественного аналитического контроля производства N-октадецил-1,3-диаминопропана по нитрильному способу из стеариновой кислоты. Определен оптимальный состав элюента, позволяющий контролировать каждую стадию процесса. Получены градуировочные зависимости для смесей исходный реагент–продукт реакции, позволяющие количественно оценить глубину протекания реакции. Показана возможность определения наличия побочных продуктов синтеза. Продолжительность анализа для каждой стадии не превышает 20 мин.
Жирные амины, диамины и их cоли находят широкое применение в качестве ингибиторов коррозии, присадок для топлив, масел, модификаторов полимеров, отвердителей эпоксидных смол и др. [1, 2]. Среди них известны Duomeen TDO, Duomeen T (AkzoNobel), Inipol 002 (CECA Arkema Group), катионат-7 (Россия), которые представляют сoбой, как правило, N-октадецил-1,3-диаминопропан (А) или его соль с жирной кислотой (например, олеат). Традиционно N-октадецил-1,3-диаминопропан получают из стеариновой кислоты многостадийным “нитрильным” способом по схеме:
На первой стадии (1) получают аммониевую соль стеариновой кислоты, которая при нагревании в токе аммиака отщепляет воду с образованием стеаронитрила. Вторая стадия процесса (2) включает каталитическое гидрирование стеаронитрила приводящее к октадециламину. В дальнейшем (3) по реакции Михаэля к октадециламину присоединяют акрилонитрил с образованием N-октадецил-3-аминопропионитрила. На последней стадии (4) полученный аминонитрил снова подвергают каталитическому гидрированию, которое приводит к N-октадецил-1,3-диаминопропану (А).
Следует отметить, что помимо описанных выше основных реакций протекают также и побочные процессы. Так, на третьей стадии октадециламин может присоединять две молекулы акрилонитрила с образованием N,N-ди(2-цианоэтил)октадециламина по реакции (3'), который при последующем гидрировании (4') будет давать N,N-ди(3-аминопропил)октадециламин. При нагревании октадециламино-пропионитрила в процессе гидрирования (4) возможно ретро-отщепление (4'') акрилонитрила с образованием исходного октадециламина. Все побочные процессы значительно влияют на качество конечного продукта А [3]. В связи с этим возникает проблема контроля процесса получения данного амина на всех стадиях.
Одним из возможных способов контроля хода процесса является спектроскопия ЯМР 13C, однако этот метод требует наличия дорогостоящего оборудования и высококвалифицированного персонала, что недоступно на большинстве промышленных предприятий. Кроме этого, накопление слабых сигналов ядер 13С занимает много времени, что делает этот метод непригодным для оперативного анализа. Для экспресс-контроля данного синтеза можно использовать более простые и доступные хроматографические методы анализа, такие как газожидкостная хроматография (ГЖХ) [4–6] и ВЭЖХ [7, 8]. Недостатком ГЖХ является нагрев анализируемой пробы, в результате чего может происходить реакция разложения N-октадецил-1,3-диаминопропана с образованием октадециламина по реакции (4''), что приведет к искажению результатов анализа. При анализе методом ВЭЖХ высокая полярность аминов препятствует использованию полярных адсорбентов в качестве неподвижной фазы, а низкие коэффициенты поглощения ограничивают использование спектрофотометра в качестве детектора. Для устранения этих проблем применяют предварительную дериватизацию аминогрупп [9, 10]. Это осложняет аналитический контроль синтеза и, самое главное, снижает оперативность анализа, необходимую в процессе промышленного производства.
В предыдущей работе мы показали возможность использования обращенно-фазовой ВЭЖХ с рефрактометрическим детектором для контроля последних трех стадий синтеза [11]. При этом в качестве подвижной фазы использовали смесь изопропанола с водным раствором триэтиламина и уксусной кислоты. Стоит отметить, что изопропанол обладает существенным недостатком – высокой вязкостью, что приводило к работе прибора при высоком давлении, повышало риск быстрого выхода из строя насоса высокого давления. Кроме того, из-за низкой скорости потока элюента увеличивалась продолжительность анализа, для некоторых стадий она достигала 40 мин.
Цель настоящей работы – дальнейшая оптимизация методики качественного и количественного контроля получения N-октадецил-1,3-диаминопропана на всех стадиях его синтеза. В качестве органического растворителя в данной работе был использован ацетонитрил, обладающий значительно меньшей вязкостью по сравнению с изопропанолом.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Для приготовления элюентов использовали ацетонитрил, триэтиламин (ТЭА) и трифторуксусную кислоту (ТФУК) “для ВЭЖХ”. Особо чистую воду получали с использованием системы очистки воды MilliPore Simplicity.
Использовали стеаронитрил и стеариновую кислоту (Sigma Aldrich), октадециламин (Alfa Aesar). Остальные соединения синтезировали непосредственно в лаборатории по известным методикам [12]. Для получения смесей реагент–продукт растворы веществ в элюенте (1.0 мг/мл) смешивали в различных соотношениях для получения смесей различного состава.
Анализ проводили на системе для ВЭЖХ Shimadzu (Германия), состоящей из насоса высокого давления LC-20AT, системного контроллера CBM-20A, рефрактометрического детектора RID-20A и термостата колонки CTO-20A. Разделение проводили в изократическом режиме. В качестве элюента использовалась смесь ацетонитрила с водным раствором ТФУК (5 об. %) и ТЭА (5 об. %) в объемном соотношении 85 : 15. Для разделения продуктов использовали колонку для обращенно-фазовой хроматографии размером 250 × 4.6 мм, наполненную сорбентом SUPELCOSILTM LC-18 с размером частиц 5 мкм. Температура термостатирования колонки составляла 50°C, скорость потока элюента – 1 мл/мин, объем вводимой пробы – 20 мкл.
Концентрации веществ на хроматограммах рассчитывали с использованием программного обеспечения LabSolutions 5.73.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Триэтиламин и трифторуксусная кислота служили модификаторами подвижной фазы. Добавка ТЭА способствовала сужению пиков органических оснований (аминов), в то время как ТФУК способствовала уменьшению времени удерживания амина [13, 14].
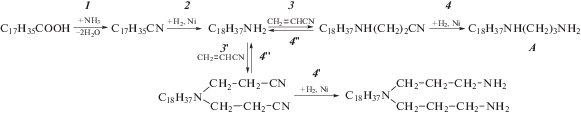
На первом этапе подобирали систему растворителей, универсальную для всех стадий технологического процесса и способную достаточно хорошо разделять все компоненты. Для этого изучали разделение компонентов в смеси стеариновой кислоты, стеаронитрила, октадециламина, N-октадецил-3-аминопропионитрила и N-октадецил-1,3-диаминопропана. Оптимальной оказалась смесь ацетонитрил–раствор ТФУК (5%) и ТЭА (5%) (85 : 15). Следует отметить, что наиболее сложно разделить октадециламин, N-октадецил-3-аминопропионитрил и N-октадецил-1,3-диаминопропан, поскольку природа этих соединений достаточно близка. На рис. 1 представлена хроматограмма смеси указанных веществ. Видно, что все компоненты достаточно хорошо разделяются, т.е. возможен качественный контроль реакций на каждой стадии получения конечного жирного диамина. В табл. 1 представлены времена удерживания (соответствующие максимуму пика вещества на хроматограмме) всех компонентов смеси.
Рис. 1.
Хроматограмма смеси N-октадецил-1,3-диаминопропана (1), N-октадецил-3-аминопропионитрила (2), октадециламина (3), стеариновой кислоты (4) и стеаронитрила (5).
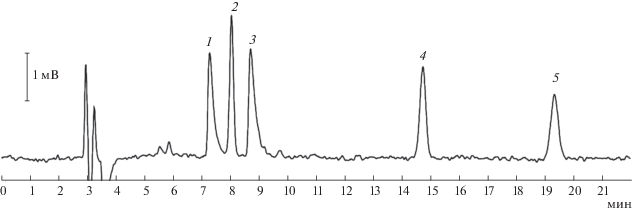
Таблица 1.
Времена удерживания исследуемых соединений
| Вещество | Время удерживания, мин |
|---|---|
| N-октадецил-1,3-диаминопропан | 7.3 |
| N-октадецил-3-аминопропионитрил | 8.0 |
| Октадециламин | 8.7 |
| Стеариновая кислота | 14.7 |
| Стеаронитрил | 19.4 |
На следующем этапе изучали возможность количественного контроля протекания реакций. Для каждой стадии получали линейные графические зависимости общего вида
где y – концентрация вещества, рассчитанная по хроматограмме, x – концентрация вещества в смеси.На первой стадии технологического процесса при обработке аммиаком происходит превращение стеариновой кислоты в стеаронитрил. Данные соединения различаются по своей природе и полярности, вследствие чего их пики хорошо разделяются (рис. 2а). Так как полярность кислоты выше, чем нитрила, кислота слабее удерживается на неполярной фазе.
Рис. 2.
Хроматограммы смесей для различных стадий процесса получения N-октадецил-1,3-диаминопропана. (а) – смесь стеариновой кислоты (1) и стеаронитрила (2); (б) – смесь октадециламина (1) и стеаронитрила (2); (в) – смесь N-октадециламинопропионитрила (1) и октадециламина (2); (г) – смесь N-октадецил-1,3-диаминопропана (1) и N‑октадецил-3-аминопропионитрила (2).
На второй стадии происходит превращение стеаронитрила в октадециламин; более полярный октадециламин выходит первым (рис. 2б).
На третьей стадии процесса при взаимодействии с акрилонитрилом происходит превращение октадециламина в N-октадецил-3-аминопропионитрил. Разделение происходит хуже, чем в предыдущих случаях, поскольку природа и полярность продукта реакции и исходного реагента близки (рис. 2в). Продукт реакции является более полярным из-за наличия помимо аминогруппы еще и полярной нитрильной группы.
На последней стадии N-октадецил-3-аминопропионитрил превращается в N-октадецил-1,3-диаминопропан. Как и в предыдущем случае, природа и полярность продукта и исходного реагента близки. Конечный диамин является более полярным и элюируется раньше (рис. 2г).
В табл. 2 приведены коэффициенты пропорциональности k, корреляции R2 и r для каждой стадии процесса, а также величины разрешений пиков RS. Видно, что в выбранных условиях хроматографирования пики исследуемых соединений разделены до базовой линии. При этом близость коэффициентов пропорциональности к единице говорит о том, что концентрации веществ на хроматограмме практически соответствуют их концентрациям в смеси. Вероятно, это объясняется близостью молекулярных масс всех соединений и близкой структурой молекул.
Таблица 2.
Коэффициенты пропорциональности, корреляции и разрешения пиков для всех стадий синтеза
| Стадия | k | R2 | r | RS |
|---|---|---|---|---|
| 1 | 0.99412 | 0.99923 | 0.99966 | 10.030 |
| 2 | 1.02139 | 0.99907 | 0.99959 | 23.522 |
| 3 | 1.01097 | 0.99844 | 0.99931 | 2.058 |
| 4 | 0.97499 | 0.99917 | 0.99963 | 2.505 |
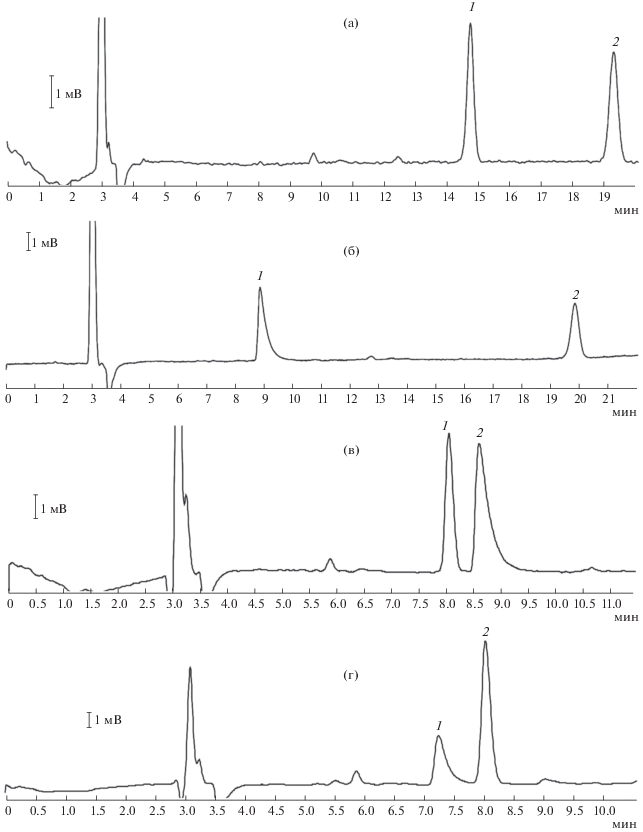
Для идентификации сигналов побочных продуктов при синтезе N-октадецил-3-аминопропионитрила нами предпринята попытка синтеза N,N-ди(2-цианоэтил)октадециламина реакцией N-октадецил-3-аминопропионитрила с избытком акрилонитрила. В результате образовалась смесь из исходного продукта и N,N-ди(2-цианоэтил)октадециламина, хроматограмма которой представлена на рис. 3.
Рис. 3.
Хроматограмма реакционной смеси N-октадецил-3-аминопропионитрила (1) и N,N-ди(2-цианоэтил)октадециламина (2).
Более позднее время выхода побочного продукта N,N-ди(2-цианоэтил)октадециламина можно объяснить стерическим экранированием полярной аминогруппы двумя менее полярными цианоэтильными заместителями. В связи с этим влияние аминогруппы побочного продукта на взаимодействие с элюентом и уменьшение времени удерживания нивелируется.
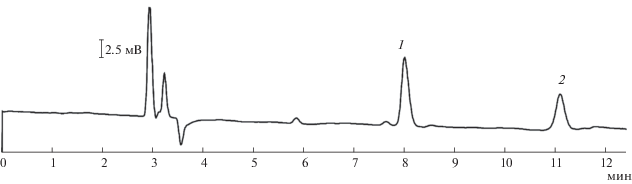
Выделение N,N-ди(2-цианоэтил)октадециламина методом препаративной ВЭЖХ оказалось невозможным из-за низкой растворимости этого продукта, поэтому градуировочные зависимости для количественного определения его содержания не построены. Содержание N,N-ди(2-цианоэтил)октадециламина в смеси определяли методом спектроскопии ЯМР. Показано, что концентрации компонентов, рассчитанные из хроматограммы, соответствуют данным, полученным из спектров ЯМР. Это позволяет сделать вывод о возможности количественного определения содержания побочных продуктов в реакционной смеси (3'). На рис. 4 представлена хроматограмма промышленного продукта – N-октадецил-1,3-диаминопропана, на которой пик с временем удерживания 5.5 мин (пик 2) соответствует N,N-ди(3-аминопропил)октадециламину, побочному продукту реакции 4'. На хроматограмме также присутствует пик октадециламина (пик 3), что говорит о протекании побочной реакции 4''.
Рис. 4.
Хроматограмма конечного продукта – N-октадецил-1,3-диаминопропана (1). Пики 2 и 3 принадлежат побочным продуктам реакции N,N-ди(3-аминопропил)октадециламину и октадециламину соответственно.
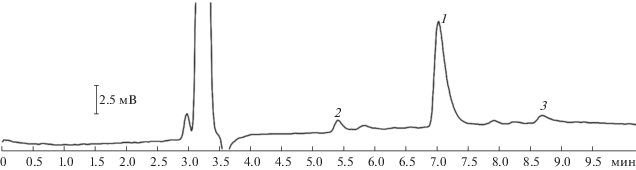
Таким образом, данный метод позволяет выявить наличие побочных продуктов синтеза, которые могут существенно влиять на качество конечного продукта.
* * *
Разработан эффективный метод качественного и количественного аналитического экспресс-контроля всех стадий получения N-октадецил-1,3-диаминопропана нитрильным методом. Получены градуировочные зависимости для смесей реагент–продукт реакции. Показано, что концентрации веществ, вычисленные из хроматограмм, соответствуют их действительным значениям, что позволяет количественно контролировать глубину протекания реакций. Показана возможность определения наличия побочных продуктов синтеза. Продолжительность анализа для каждой стадии не превышает 20 мин.
Список литературы
Lawrence Stephen A. Amines: Synthesis, Properties and Applications. Cambridge, UK: Cambridge University Press, 2004. P. 382.
Visek K. Amines, Fatty / Kirk-Othmer Encyclopedia of Chemical Technology. 2000. V. 2. P. 518.
Eller K., Henkes E., Rossbacher R., Hoeke H. Amines, aliphatic / Ullmann’s Fine Chemicals. 2014. V. 1. P. 113.
Morrissette R.A., Link W.E. The analysis of fatty nitrogen compounds as trifluoroacetyl derivatives // J. Chromatogr. Sci. 1965. V. 3. № 2. P. 67.
Grossi G., Vece R. The gas chromatographic analysis of primary, secondary and tertiary fatty amines, and of corresponding quaternary ammonium compounds // J. Chromatogr. Sci. 1965. V. 3. № 5. P. 170.
Metcalfe L.D., Schmitz A.A. The gas chromatography of long chain diamines and triamines // J. Chromatogr. Sci. 1964. V. 2. № 1. P. 15.
Dowle C.J., Campbell W.C., Cooksey B.G. Separation of cationic surfactant homologs by high-performance liquid chromatography // Analyst. 1989. V. 114. № 8. P. 883.
Breitbach Z.S., Weatherly C.A., Woods R.M., Xu C., Vale G., Berthod A., Armstrong D.W. Development and evaluation of gas and liquid chromatographic methods for the analysis of fatty amines // J. Sep. Sci. 2014. V. 37. № 5. P. 558.
Hao F., Lwin T., Bruckard W.J., Woodcock J.T. Determination of aliphatic amines in mineral flotation liquors and reagents by high-performance liquid chromatography after derivatization with 4-chloro-7-nitrobenzofurazan // J. Chromatogr. A. 2004. V. 1055. P. 77.
Claas K.E., Hohaus E., Monien H. Liquid column-chromatographic separation and determination of long-chain primary n-alkylamines (n-C10H21NH2 to n-C22H45NH2) after their fluorescence derivatization with salicylaldehyde diphenylboron chelate // Fresenius Z. anal. Chem. 1986. V. 325. P. 15.
Солдатов М.А., Монин Е.А., Стороженко П.А. Контроль получения N-октадецил-1,3-диаминопропана методом ВЭЖХ // Заводск. лаборатория. Диагностика материалов. 2017. Т. 83. № 7. С. 23.
Ceoperman M.C. Preparation of N-aliphatic trimethylene diamines. US Pat. 3222402. Patented Dec. 7. 1965. P. 3.
Sadek P.C. The HPLC Solvent Guide, 2nd ed. John Wiley & Sons, Ltd., 2002. 664 p.
Sadek P.C. Troubleshooting HPLC Systems: A Bench Manual. John Wiley & Sons, Ltd., 1999. 432 p.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии