БИОФИЗИКА, 2023, том 68, № 5, с. 920-925
МОЛЕКУЛЯРНАЯ БИОФИЗИКА
УДК 577.3
ПОИСК АНОМАЛИЙ СИГНАЛА ПОКРЫТИЯ СЕКВЕНИРОВАНИЯ,
АССОЦИИРОВАННЫХ СО СТРУКТУРНЫМИ ВАРИАЦИЯМИ ГЕНОМА
© 2023 г. И.В. Бездворных*, Н.А. Черкасов*, А.А. Канапин*, А.А. Самсонова*, #
*Санкт-Петербургскии государственный университет,
Университетская набережная, 7-9, Санкт-Петербург, 199034, Россия
#E-mail: a.samsonova@spbu.ru
Поступила в редакцию 04.10.2022 г.
После доработки 04.10.2022 г.
Принята к публикации 25.10.2022 г.
Структурные вариации генома являются одним из основных источников генетического разнообра-
зия. Как мутагены структурные варианты могут оказывать значительное влияние на здоровье чело-
века, являясь причинами наследственных и онкологических заболеваний. Существующие методы
поиска структурных вариантов основываются на анализе данных высокопроизводительного секве-
нирования, и, несмотря на значительный прогресс в их развитии, не позволяют определять струк-
турные вариации с точностью, достаточной для применения в диагностике. Новые возможности
для разработки методов поиска структурных вариаций представляет анализ сигнала покрытия се-
квенирования (т.е. количества фрагментов секвенирования, выравненных в каждой точке генома),
который может рассматриваться как временной ряд. В работе представлен метод для поиска повто-
ряющихся паттернов в сигнале покрытия, разработанный с использованием алгоритмов, применя-
емых для анализа временных рядов, а именно: KNN- (K-nearest neighbour) и SAX-преобразования
(Symbolic Aggregation Approximation) сигнала. С использованием данных проекта Human Genome
Diversity Project, включающих полногеномное секвенирование 911 человек разного этнического
происхождения, нами были построены обобщающие паттерны сигнала покрытия в окрестностях
точек разрыва, соответствующих структурным вариациям. В дополнение был разработан программ-
ный пакет для быстрого поиска аномалий в сигнале покрытия с применением полученных пат-
тернов.
Ключевые слова: геномика, секвенирование, структурные вариации, анализ сигналов, SAX-трансфор-
мация.
DOI: 10.31857/S0006302923050113, EDN: PHCXCT
of coverage) и анализ расщепленных фрагментов
Структурные вариации включают в себя такие
масштабные перестройки генома, как протяжен-
секвенирования (split reads) [4].
ные делеции, инсерции, транслокации, инверсии,
Несмотря на значительный прогресс, достигну-
а также изменения уровня копийности (copy num-
тый в области разработки способов обнаружения
ber variation, CNV) [1]. В отличие от однонуклео-
структурных вариантов, задача точной их локали-
тидных замен и коротких (менее 30 п.о.) инсерций
зации до сих пор не имеет удовлетворительного ре-
и делеций, структурные вариации, в силу своего
шения, что выражается в значительном уровне
размера, могут оказывать значительное влияние на
разногласий в результатах поиска [5, 6]. Иначе го-
фенотип организма хозяина и, в некоторых случа-
воря, цель, заключающаяся в нахождении точки
ях, провоцировать возникновение заболеваний и
разрыва с точностью в плюс/минус одно основа-
наследственных патологий [2, 3]. Современные
ние, необходимая для применения результатов в
алгоритмы поиска структурных вариаций базиру-
клинической практике не достигнута. Одним из
ются на использовании данных высокопроизводи-
способов решения проблемы неточности обнару-
тельного секвенирования (полногеномного или
жения координат структурных вариаций в геноме,
полноэкзомного) и используют две основных ме-
является разработка алгоритмов, анализирующих
тодологии: анализ уровня сигнала покрытия (depth
и интегрирующих результаты предсказания раз-
Сокращение: SAX - аппроксимация с помощью символь-
личных программ (так называемых meta-callers),
ной агрегации (Symbolic Aggregation Approximation).
таких, как svclassify и подобных [7].
920
ПОИСК АНОМАЛИЙ СИГНАЛА ПОКРЫТИЯ СЕКВЕНИРОВАНИЯ
921
В то же время анализ уровня сигнала покрытия
ваны в соответствии с генотипом (гомо- или гете-
с точки зрения поиска аномалий и аберраций
розигота) структурных вариантов, что позволило
изучен относительно мало, и лишь немногие про-
в конечном итоге получить 22 группы для поиска
граммные пакеты используют этот источник ин-
устойчивых паттернов.
формации о форме, протяженности, амплитуде,
SAX-преобразование и модифицированный
тренде и пр. сигнала для поиска структурных ва-
алгоритм KNN. Aппроксимация с помощью
риантов [8, 9]. Под сигналом покрытия генома
символьной агрегации позволяет осуществить
понимается вектор целочисленных значений,
преобразование временного ряда, существенно
значение которого в каждой точке соответствует
снижающее размерность входных данных. Клас-
числу выровненных на референсный геном фраг-
сификаторы, построенные на преобразованных
ментов секвенирования, покрывающих данную
данных, обладают обобщающей способностью
позицию. Такой сигнал может рассматриваться
без ущерба для качества классификации [10]. Для
как временной ряд и, следовательно, к нему могут
данного преобразования требуется определить
быть применены разнообразные алгоритмы и ме-
два параметра — ширину окна W и размер алфа-
тоды, изначально разработанные для анализа
вита A. Преобразование включает в себя следую-
временных рядов и, например, поиска разладок и
щие этапы:
характеристических паттернов (так называемых
1. Нормализация исходного сигнала на сред-
мотивов) в них.
нее и дисперсию.
В данной работе мы представляем новый ме-
2. Для заданной длины алфавита A генерирует-
тод поиска устойчивых паттернов, ассоциирован-
ся интервальная сетка, где значения распределе-
ных с точками разрыва, соответствующих различ-
ны по нормальному закону. Каждое i-e значение
ным типам структурных вариаций, разработан-
сетки является i/A-квантилем функции плотно-
ный с использованием алгоритма KNN_Search
сти нормального распределения.
(K-nearest neighbours) [10] и аппроксимации с по-
3. Сигнал разбивается на W непересекающих-
мощью символьной агрегации (Symbolic Aggrega-
ся одинаковых сегментов и значения в каждом
tion Approximation, SAX) [11]. На основе анализа
сегменте усредняются.
данных, содержащихся в ранее разработанной
нами базе данных SWaveform [12], были построе-
4. Каждое усредненное значение сегмента
ны устойчивые повторяющиеся паттерны (моти-
округляется до ближайшего значения из сетки из
вы) для имеющихся типов структурных вариаций.
шага 2. Этому сегменту присваивается соответ-
Также нами был разработан инструмент быстрого
ствующий номер сетки (от 0 до A-1).
измерения SAX-расстояния между профилем по-
Метод поиска K-ближайших соседей (K-Near-
крытия генома и найденными нашим алгоритмом
est Neighbour, KNN_Search) для заданного векто-
(или любыми другими) мотивами, позволяющий
ра v, заданного максимального расстояния T и из-
обнаруживать аберрантные регионы в сигнале
вестной функции расстояния ρ, возвращает спи-
покрытия, потенциально связанные со структур-
сок векторов, которые находятся на расстоянии
ными вариациями.
от v не далее, чем на T. При анализе сигнала по-
крытия, последний рассматривается как одно-
мерный временной ряд и разбивается скользя-
МЕТОДЫ
щим окном на векторы, каждый из которых пре-
Источники данных. База данных SWaveform
образуется в свое SAX-представление {v0, …, vV}.
[12] содержит профили сигнала покрытия секве-
Между этими векторами рассчитываются попар-
нирования, полученные на основании анализа
ные расстояния. Из них 1.5% самых малых рас-
данных полногеномного секвенирования 911 об-
стояний между векторами считаются максималь-
разцов консорциума Human Genome Diversity
ным значением T для алгоритма KNN_Search.
Project [13, 14] со средним значением уровня по-
Все векторы, которые оказались ближе друг к
крытия 30x, общее число профилей составляет
другу, чем T, объединяются в группы, которые
13106216. Для всех имеющихся в базе данных ти-
сортируются по убыванию числа элементов в них.
пов структурных вариаций (делеции, дуплика-
Таким образом, группа, содержащая максималь-
ции, инверсии, инсерции и варианты копийно-
ное число объединенных векторов, соответствует
сти) нами были отобраны группы профилей сиг-
преобладающему мотиву.
нала покрытия в окне ±256 п.о. от точки разрыва
Поскольку входящий временной ряд может
(breakpoint). Для анализа были использованы
оказаться достаточно длинным, возникает доста-
только варианты с длиной, большей размера ок-
точно ресурсоемкая задача измерения попарных
на, т.е. превышающих 512 п.о. В случае протяжен-
SAX-расстояний между всеми векторами {v0, …,
ных вариаций, а именно делеций, дупликаций,
инверсий и вариантов копийности, использова-
v
V}. Чтобы существенно снизить число операций
лись профили, соответствующие их левой и пра-
по измерению расстояния, нами была произведе-
вой границам. Также профили были сгруппиро-
на следующая модификация исходного алгорит-
БИОФИЗИКА том 68
№ 5
2023
922
БЕЗДВОРНЫХ и др.
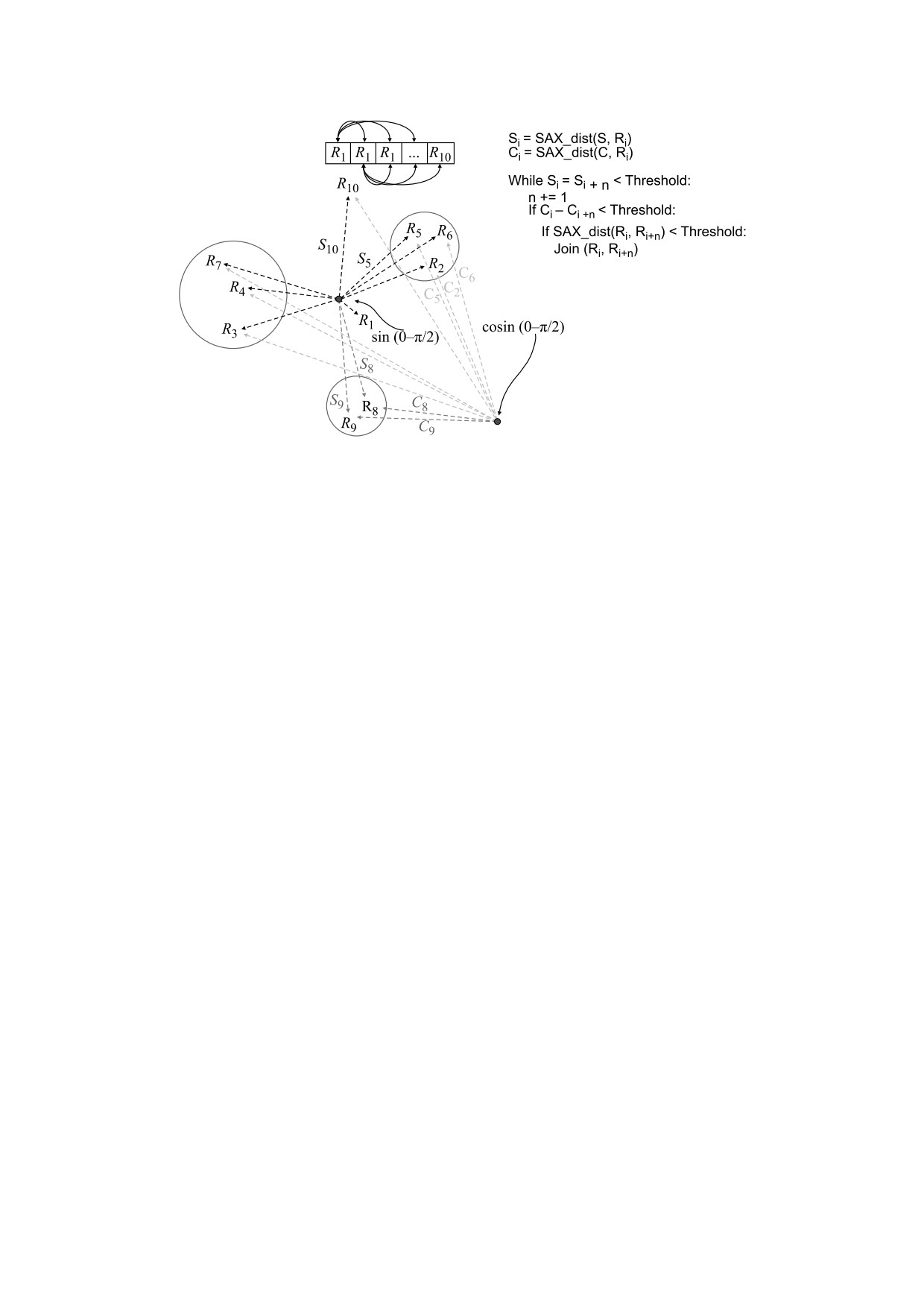
Рис. 1. Схематическое представление модифицированного алгоритма KNN. Расстояния от базисных векторов S и C
до всех остальных векторов - Si и Ci. Расстояние между любыми двумя векторами R может быть меньше порогового
тогда и только тогда, когда разница их расстояний до каждого из референсных векторов меньше порогового.
ма KNN_Search. На первом этапе производится
комых групп рассчитывали силуэтный индекс,
предварительный подсчет расстояний от произ-
который послужил критерием выбора итогового
вольно выбранных нетождественных референс-
разбиения. Для оценки расстояния между профи-
ных векторов {b0, …, bB} до всех векторов списка
лями сигнала был использован метод расчета, ос-
{v0, …, vV}. Затем, для каждой пары векторов vi и vj
нованный на принципе динамической трансфор-
мации временной шкалы (Dynamic Time Warping,
проверяются расстояния до референсных векто-
DTW) [15-17]. Существенной его особенностью
ров: если существует хотя бы один референсный
является устойчивость к сдвигу расстояния между
вектор bk, такой, что |ρ(vi,bk) - ρ(vj,bk)| > T, то, со-
сравниваемыми сигналами, что позволяет срав-
гласно положениям, представленным в работе
нивать профили, в которых общий паттерн сиг-
[10], ρ(vi,vj)| > T, т.е. расстояние между данной па-
нала сохраняется, но существуют локальные ис-
рой векторов vi и vj также больше порогового.
кажения формы (т.е. растяжения или сжатия).
Схематически данный метод показан на рис. 1.
Применение этого метода абсолютно оправдано в
Такая модификация исходного алгоритма позво-
случае оценки расстояния между интервалами,
ляет существенно сократить количество опера-
содержащими точки разрыва, порожденные
ций вычисления попарных расстояний. Вопрос
структурными вариантами, так как дают возмож-
выбора референсных векторов {b0, …, bB} остается
ность компенсировать эффекты, вызванные не-
открытым, однако с математической точки зре-
точностями в работе алгоритмов предсказания
ния их выбор может быть произвольным, поэто-
структурных вариаций. В зависимости от класса
му в нашей модификации алгоритма используют-
структурного варианта алгоритм выделяет, как
ся два референсных вектора - SAX-трансформа-
правило, либо один преобладающий кластер (т.е.
ции функций sin и cos в диапазоне от 0 до π/2.
содержащий более 2/3 профилей сигнала) или же
два примерно равных по размерам кластера. В
Построение повторяющихся паттернов (моти-
случае наличия преобладающего кластера наи-
вов) на основе профилей сигнала покрытия. Поиск
меньший исключается из дальнейшего анализа.
паттернов для каждой из 22 групп профилей сиг-
нала покрытия, описанных выше, проводили сле-
На следующем этапе все профили подвергают-
дующим образом. Исходные профили группиро-
ся SAX-преобразованию, где размер алфавита со-
вались с применением алгоритма кластеризации
ставляет 24 символа, а число сегментов выбрано
K-means с числом кластеров равным 2. Число k,
равным 64. На завершающем этапе внутри полу-
определяющее количество кластеров, оптималь-
ченного кластера или кластеров производится
ное применительно к данной задаче, было опре-
поиск мотива, преобладающего в полученных
делено в ходе серии вычислительных экспери-
SAX-трансформированных профилях с исполь-
ментов с выборкой профилей, соответствующих
зованием описанного выше модифицированного
структурным вариантам одного класса и геноти-
алгоритма KNN. Итоговый мотив сохраняется в
па. В ходе кластеризации с различным числом ис-
виде вектора усредненных значений для каждой
БИОФИЗИКА том 68
№ 5
2023
ПОИСК АНОМАЛИЙ СИГНАЛА ПОКРЫТИЯ СЕКВЕНИРОВАНИЯ
923
позиции. Также для каждого мотива сохраняются
тивов, ассоциированных с вышеуказанными ти-
параметры SAX-преобразования.
пами структурных вариаций, предсказанных с
использованием данных ресурса SWaveform. Мы
Использование SAX-мотивов для поиска анома-
исходили из того, что мотив может встречаться в
лий в сигнале покрытия. Для поиска участков ге-
геноме не только в позициях, идентифицирован-
нома, в которых сигнал покрытия обладает мак-
ных DELLY2, но и в других, поэтому для нас было
симальным сходством с полученными SAX-моти-
вами нами был разработан программный
критически важным увеличить чувствительность,
но не так существенно снизить специфичность
нахождения точек разрыва. Регионы, обнаружен-
реализованный на языках C и Python и способ-
ные в ходе анализа с помощью описанного пакета
ный работать в многопоточном режиме. Входны-
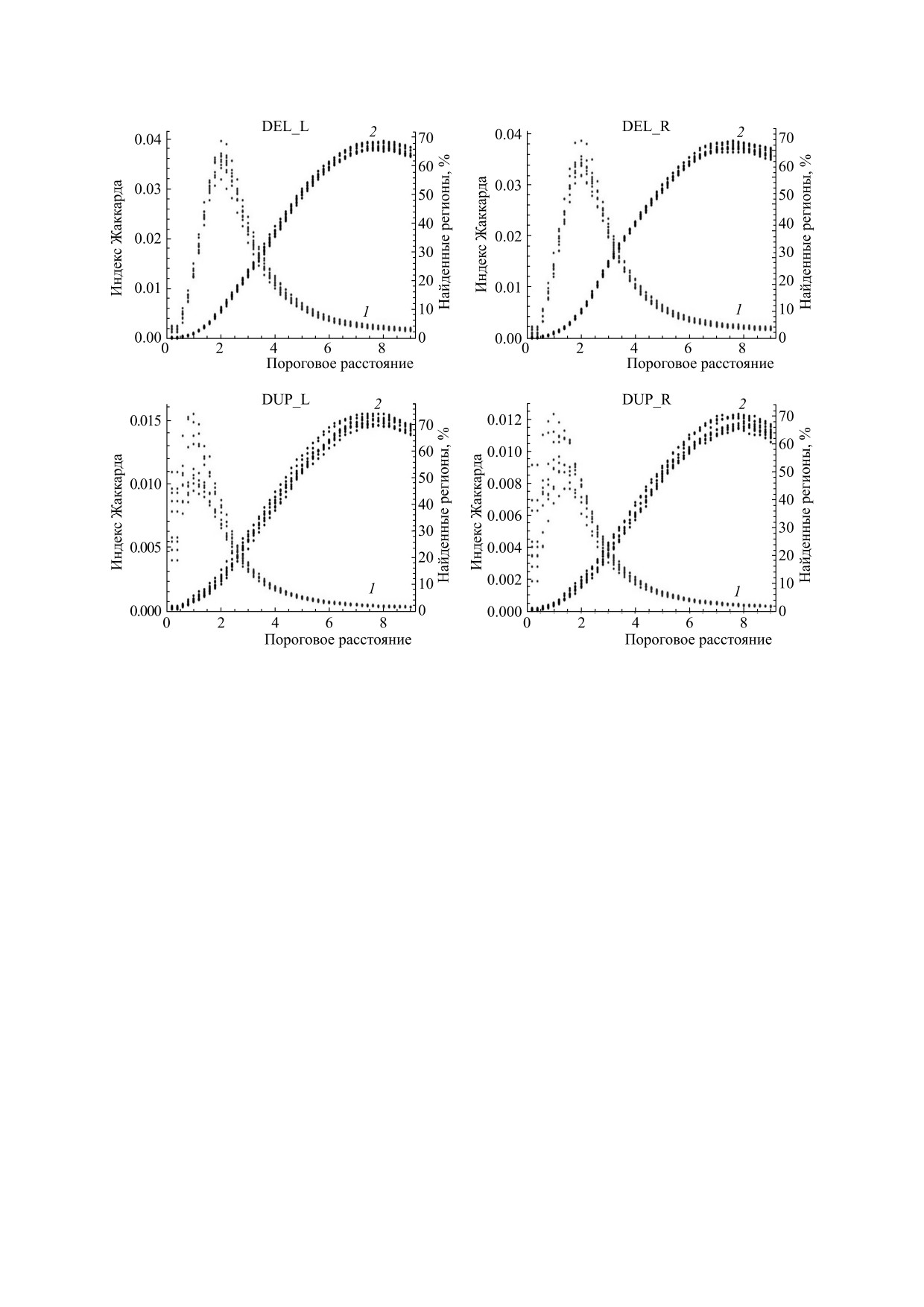
saxmf, были отфильтрованы по пороговому зна-
ми данными для работы программы является сиг-
нал покрытия в специальном формате BCOV [12],
чению SAX-расстояния, изменявшегося от 0 до
10 с шагом 0.2. Для каждого случая рассчитывали
получаемый путем преобразования стандартных
расстояние Жаккарда между регионами, найден-
файлов выравнивания фрагментов секвенирова-
ными saxmf и DELLY2, а также процент локусов,
ния в форматах SAM/BAM/CRAM. Преобразова-
найденных только с помощью DELLY2. Результа-
ние осуществляется с помощью программ mos-
depth
[18] и bed2cov (см. репозиторий
ты анализа демонстрируют высокий уровень сов-
падения результатов поиска saxmf с данными, по-
лученными одной из самых распространенных
грамма saxmf включает в себя SAX-мотивы, рас-
программ для поиска структурных вариаций (см.
считанные из данных Human Genome Diversity
рис. 2).
Project, однако имеется возможность поиска лю-
бых других мотивов, закодированных в формате
Таким образом, можно предварительно оце-
JSON.
нить пороговое значение SAX-расстояния для
В ходе работы программы сигнал покрытия
фильтрации результатов последующего анализа
подвергается SAX-трансформации в скользящем
данных с помощью описанного метода.
окне, после чего вычисляется SAX-расстояние
В дополнение был проведен поиск регионов,
между ним и искомым мотивом. Полученные в
содержащих паттерн, ассоциированный с левой
ходе первичного поиска геномные интервалы,
границей делеций в эталонном образце генома
потенциально содержащие мотив, соответствую-
HG002 [21, 22] (проект Genome In a Bottle [23]
щий точке разрыва, порожденной каким-нибудь
(GIAB), NIST). Под эгидой консорциума GIAB в
структурным вариантом, на следующем этапе,
этом геноме был проведен всесторонний анализ
объединяются в кластеры для идентификации ре-
структурных вариаций с использованием различ-
гиона, имеющего максимальную близость с иско-
ных технологий секвенирования, включающих
мым мотивом. Положение центральной точки
использование длинных и коротких фрагментов,
данного региона ищется как взвешенное среднее
а также программ для предсказания структурных
по SAX-расстоянию по формуле:
вариаций. Полученные результаты были проана-
лизированы кураторами, что позволило охаракте-
i
Pos *SAX
i
ризовать наиболее достоверные вариации [24].
i
BND =
,
В частности, для выбранного нами мотива (левая
(S-SAX
)
i
граница делеций) в данном геноме описано
i
5372 региона, из которых нами был успешно об-
где S - максимальное SAX-расстояние внутри
наружен 4651 регион (~86%). Этот результат по-
кластера, Posi — центр i-го региона, SAXi - SAX-
казывает, что разработанная методика поиска
расстояние i-го региона до мотива. Результаты
паттернов сигнала покрытия может быть успеш-
поиска выводятся в формате BED-файла, вклю-
но применена для первичного поиска структур-
чающего координаты региона и SAX-расстояние
ных вариантов в данных полногеномного секве-
до искомого мотива.
нирования.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
ФИНАНСИРОВАНИЕ РАБОТЫ
Проверка предложенного метода поиска пат-
Работа выполнена при финансовой̆поддержке
тернов, ассоциированных со структурными вари-
Российского научного фонда (грант No 20-14-
антами, была проведна на двух независимых на-
00072).
борах данных. В первом случае были использова-
ны результаты полногеномного секвенирования
проекта «Геномы России» [19]. Для 360 образцов
КОНФЛИКТ ИНТЕРЕСОВ
были определены структурные варианты (деле-
ции и дупликации) с использованием программы
Авторы заявляют об отсутствии конфликта
DELLY2 [20]. Затем нами был проведен поиск мо-
интересов.
БИОФИЗИКА том 68
№ 5
2023
924
БЕЗДВОРНЫХ и др.
Рис. 2. Графики зависимости индекса Жаккарда (1) и процента найденных регионов (2) от порогового расстояния (ось
абсцисс).
СОБЛЮДЕНИЕ ЭТИЧЕСКИХ СТАНДАРТОВ
12. 762 BGRS/SB-2022 Swaveform: a genome-wide survey
of structural variation profiles, Thirteen Int. Multicon-
Настоящая работа не содержит описания ис-
ference (2022).
следований с использованием людей и животных
13. A. Bergström, et al., Science, 367 (6484), eaay5012
в качестве объектов.
(2020).
14. M. A. Almarri, et al., Cell, 182 (1), 189 (2020).
СПИСОК ЛИТЕРАТУРЫ
15. H. Sakoe and S. Chiba, IEEE Trans. Acoust. Speech
Signal Process., 26 (1), 43 (1978).
1. R. L. Collins, et al., Nature, 581 (7809), 444 (2020).
16. F. Petitjean, A. Ketterlin, and P. Gançarski, Pattern
2. Y. R. Li, et al., Nature Commun., 11 (1), 255 (2020).
Recogn., 44 (3), 678 (2011).
3. S. Girirajan, et al., Am. J. Human Genetics, 92 (2), 221
17. R. Tavenard, et al., J. Mach. Learn. Res., 21 (118), 1
(2013).
(2020).
4. M. Mahmoud, et al., Genome Biol., 20 (1), 1 (2019).
18. B. S. Pedersen and A. R. Quinlan, Bioinformatics, 34
5. S. Kosugi, et al., Genome Biol., 20 (1), 117 (2019).
(5), 867 (2018).
6. Z. Liu, et al., Genome Biol., 23 (1), 68 (2022).
19. D. V. Zhernakova, et al., Genomics, 1 (2019).
7. H. Parikh, et al., BMC Genomics, 17 (1), 64 (2016).
20. T. Rausch, et al., Bioinformatics, 28 (18), i333 (2012).
8. A. Abyzov, et al., Genome Res., 21 (6), 974 (2011).
21. J. M. Zook, et al., Nat. Biotechnol., 1 (2020).
9. M. Rapti, et al., Brief Bioinform., 23 (2), bbac049
(2022).
22. A. Shumate, et al., Genome Biol., 1 (2020).
10. Z. A. Aghbari, Data Knowl. Eng., 52 (3), 333 (2005).
23. J. M. Zook, et al., Sci. Data, 3, 160025 (2016).
11. S. Malinowski, et al., Lect. Notes Comput. Sci., 273
24. L. M. Chapman, et al., PLoS Comput. Biol. 16 (6),
(2013).
e1007933-20 (2020).
БИОФИЗИКА том 68
№ 5
2023
ПОИСК АНОМАЛИЙ СИГНАЛА ПОКРЫТИЯ СЕКВЕНИРОВАНИЯ
925
Searching for Sequencing Signal Anomalies Associated
with Genome Structural Variations
I.V. Bezdvornykh*, N.A. Cherkasov*, A.A. Kanapin*, and A.A. Samsonova*
*St. Petersburg State University, Universitetskaya nab. 7-9, St. Petersburg, 199034 Russia
Genomic structural variations (SVs) are one of the main sources of genetic diversity. Structural variants as
mutagens may have a significant impact on human health and lead to hereditary diseases and cancers. Exist-
ing methods of finding structural variants are based on analysis of high-throughput sequencing data and de-
spite significant progress in the development of the detection methods, there is still a need for improving the
identification of structural variations with accuracy appropriate for use in a diagnostic procedure. Analysis of
the signal of sequencing coverage (i.e., the number of sequencing fragments that aligned to every point of a
genome) holds new potential for the design of approaches for structural variations discovery, and can be used
as time-series analysis. Here, we present an approach for identification of patterns in the coverage signal. The
method has been developed based on algorithms used for analysis of time series data, namely KNN (K-near-
est neighbour) search algorithm and the SAX (Symbolic Aggregation Approximation) method. Using the rich
dataset encompassing full genomes of 911 individuals with different ethnic backgrounds generated by the Hu-
man Genome Diversity Project initiative, we constructed generalized patterns of signal coverage in the vicin-
ity of breakpoints corresponding to various structural variant types. Also, with the benefit of the SAX models
of the motifs we developed a software package for fast detection of anomalies in the coverage signal.
Keywords: genomics, sequencing, structural variations, signal analysis, SAX transformation
БИОФИЗИКА том 68
№ 5
2023