БИОФИЗИКА, 2023, том 68, № 5, с. 911-919
МОЛЕКУЛЯРНАЯ БИОФИЗИКА
УДК 577.22
УТОЧНЕНИЕ ПОЗИЦИЙ НУКЛЕОСОМ ВНУТРИ ОТДЕЛЬНЫХ ГЕНОВ
С ИСПОЛЬЗОВАНИЕМ МЕТОДОВ МОЛЕКУЛЯРНОГО
МОДЕЛИРОВАНИЯ И ДАННЫХ MNASE-СЕКВЕНИРОВАНИЯ
© 2023 г. В.А. Васильев*, Д.М. Рябов*, А.К. Шайтан*, Г.А. Армеев*, #
Московский государственный университет имени М.В. Ломоносова, Ленинские горы, 1/12, Москва, 119234, Россия
#E-mail: armeev@intbio.org
Поступила в редакцию 06.05.2023 г.
После доработки 06.05.2023 г.
Принята к публикации 17.05.2023 г.
Организация хроматина играет важную роль в регуляции работы генетического аппарата клетки.
Основной единицей упаковки хроматина является нуклеосома, хранящая на себе ДНК длиной
около 145 пар нуклеотидов. Упаковка генетического материала и его доступность для ферментов
транскрипции и других регуляторных хроматиновых белков зависят от позиции нуклеосом. Для
исследования позиций нуклеосом в геноме применяют MNase-секвенирование. Данные MNase-
секвенирования позволяют детектировать факт наличия нуклеосом на последовательности, однако
их точное позиционирование сложно установить по этим данным. Для уточнения положений
нуклеосом необходимо дополнительно фильтровать и обрабатывать данные. В данной работе
предлагается комбинированный метод отбора возможных позиций нуклеосом по данным MNase-
секвенирования, основанный на геометрическом анализе молекулярных моделей нуклеосомных
цепочек. Разработанный алгоритм позволяет эффективно отсеивать недоступные комбинации
нуклеосомных цепочек и конформационно запрещенные позиции нуклеосом.
Ключевые слова: хроматин, нуклеосома, позиционирование нуклеосом, молекулярное моделирование.
DOI: 10.31857/S0006302923050101, EDN: PHCUCE:
Геномы большинства эукариот содержат боль-
у пекарских дрожжей на один ген приходится
ше ДНК, чем геномы прокариот. Такое различие
около 2000 н.п. и примерно 70% генома дрожжей
можно ожидать, так как для работы более сложно
занято кодирующими последовательностями
устроенных организмов требуется большее число
белков, которых у дрожжей около 6000. Геномы
генов. Однако размер генома не связан напрямую
высших животных (например, человека) устрое-
со сложностью организма. Так, многие растения
ны еще сложнее и содержат продолжительные об-
обладают значительно большим по длине гено-
ласти некодирующей ДНК. По современным
мом, чем человек. Причина различий в размерах
оценкам только 1.5% из примерно 3 миллиардов
геномов между эукариотами и прокариотами
н.п. генома человека кодирует последовательно-
кроется в ряде причин. Эукариотические геномы
сти белков [1]. Остальные участки заняты неко-
содержат некодирующие участки ДНК, располо-
дирующими последовательностями, которые тем
женные между и внутри генов. В отличие от про-
не менее выполняют регуляторные функции. Не-
кариот, некоторые эукариотические гены много-
смотря на линейный размер, эукариотический ге-
кратно повторяются. Наличие множественных
ном помещается в клеточном ядре, при этом со-
копий генов позволяет значительно повысить
храняя свои функции.
уровни экспрессии кодируемых белков. Так, на-
Базовым элементом компактизации хромати-
пример, кодируются белки-гистоны - одни из са-
на является нуклеосома. Нуклеосомы состоят из
мых распространенных белков в клеточном ядре.
восьми гистонов (четыре типа гистонов, форми-
В геномах бактерий большая часть ДНК коди-
рующих гетеродимеры) и ДНК длиной порядка
рует белки. Например, геном E. coli состоит при-
145 пар нуклеотидов. Нуклеосомы - симметрич-
мерно из 5 миллионов н.п. и содержит около 4 ты-
ные структуры (рис. 1а), при этом ось симметрии
сяч генов, при этом почти 90% ДНК кодирует
(диадная ось нуклеосомы) в них проходит вблизи
белковые последовательности. Геном пекарских
центра одной из нуклеотидных пар (далее по тек-
дрожжей, состоящий из 12 миллионов н.п., при-
сту диадная н.п., на рис. 1а показана черной точ-
мерно в 2.5 раза больше генома E. coli. В среднем
кой). Упаковка генетического материала и его до-
911
912
ВАСИЛЬЕВ и др.
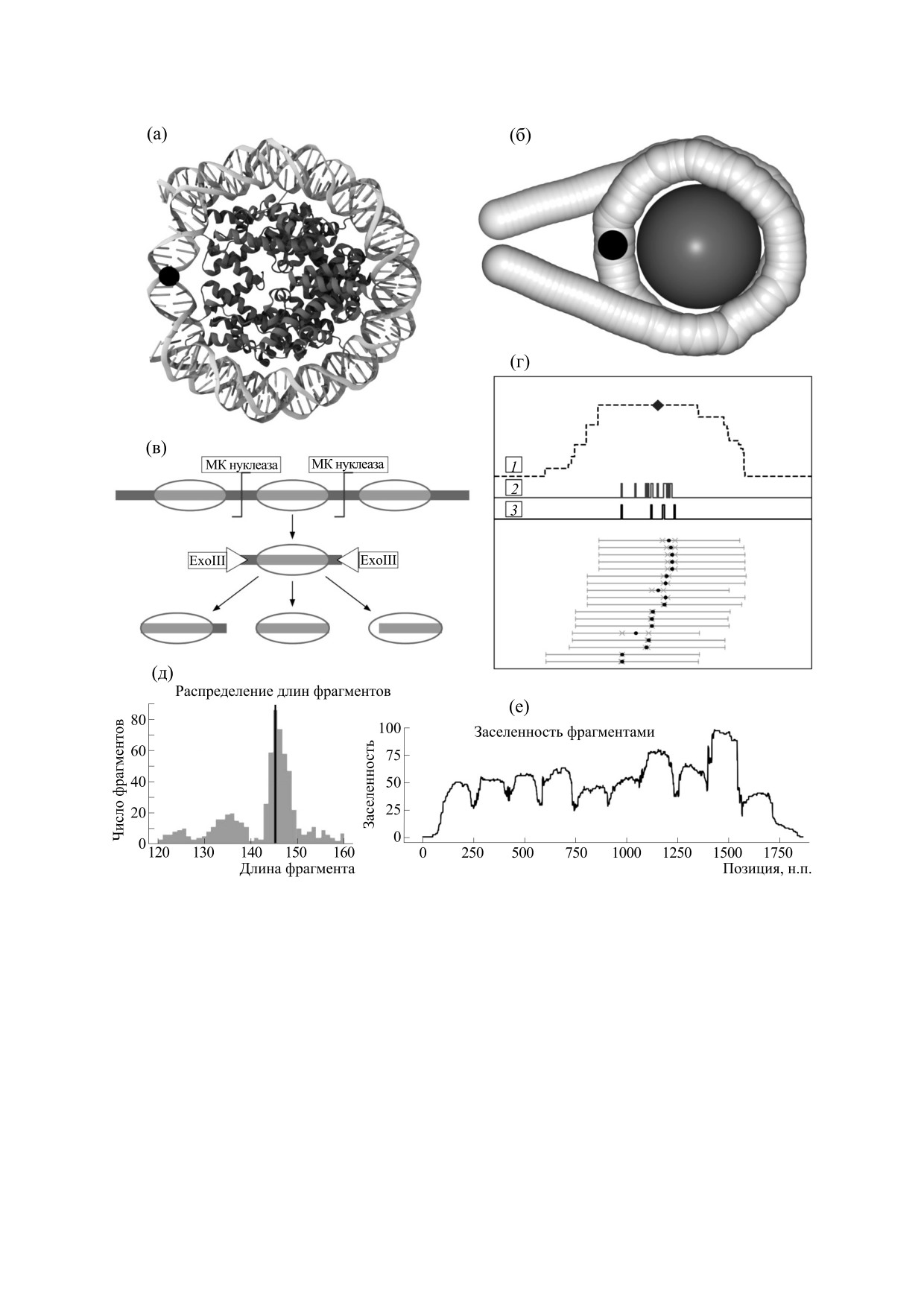
Рис. 1. (а) - Внешний вид структуры нуклеосомы. Визуализация построена на основе модели 3LZ0 из PDB. Белковая
часть показана в виде вторичной структуры темно серым цветом, ДНК показана светло-серым. Черной точкой отмечено
положение диадной н.п. (б) - Внешний вид огрубленной модели нуклеосомной частицы с линкерными областями.
Черной точкой отметено положение диадной н.п. Цветовая схема соответствует рисунку (а). (в) - Схема появления
фрагментов разной длины в экспериментах по MNase-секвенированию. (г) - Схема формирования пиков на профилях
заселенности нуклеосомами. Отрезки в нижней половине схемы отображают фрагменты нуклеосомной ДНК из MNase-
секвенирования, черные точки - положения центров фрагментов, серые крестики - возможные положения диадных
н.п. Линия 1 - сумма всех фрагментов, ромбом показано положение центра пика (данное положение часто используют
как статистическое среднее расположение нуклеосом); линия 2 - центров сигналов; линия 3 - отфильтрованный
профиль положения диадных н.п. (д) - Распределение длин выровненных участков. (е) - Профили заселенности
участков генома нуклеосомами.
ступность для ферментов транскрипции и других
ность нуклеосом влияет последовательность
регуляторных белков хроматина зависят от рас-
ДНК. Существуют такие последовательности, на
положения нуклеосом. Известно, что на стабиль-
которых нуклеосомы не только стабильны, но и
БИОФИЗИКА том 68
№ 5
2023
УТОЧНЕНИЕ ПОЗИЦИЙ НУКЛЕОСОМ ВНУТРИ ОТДЕЛЬНЫХ ГЕНОВ
913
строго позиционированы [2]. Однако фундамен-
зонуклеазой III (ExoIII), которая расщепляет не
тальные механизмы влияния последовательности
защищенные белками концы ДНК. Полученные
ДНК на стабильность и позиционирование нук-
фрагменты ДНК очищают от белков и определя-
леосом неясны. Позиционирование нуклеосом в
ют методами высокопроизводительного секвени-
хроматине обеспечивается не только сродством к
рования. Получившиеся чтения картируют на ге-
последовательности, но и работой транскрипци-
ном и фильтруют по длине. Участки длиной по-
онных факторов, которые активно передвигают
рядка 150 н.п. обычно ассоциируют с областями
нуклеосомы по ДНК (ремоделлеров хроматина).
расположения нуклеосом [10]. В ходе дальнейше-
Например, гены, на которых активен ISW1, в
го анализа рассчитывают распределения длин вы-
среднем содержат нуклеосомы каждые 175 н.п., а
равненных участков (рис. 1д) и определяют про-
гены, регулируемые CHD1, - 160 н.п.[3]
фили заселенности участков генома нуклеосома-
Нуклеосомы в геноме распределены не слу-
ми - для каждого нуклеотида определяется число
чайно. Существуют как области с высокой упоря-
сигналов от участков нуклеосомной ДНК
доченностью нуклеосом, так и участки генома,
(рис. 1е).
лишенные четко определенной нуклеосомной
Важно отметить, что полученные сигналы (вы-
организации. В работе [4] впервые было показа-
равненные на геном участки нуклеосомной ДНК)
но, что в областях вблизи промоторов S. сerevisiae
нуклеосомы точно позиционированы. Такое по-
не позволяют однозначно судить о позиции нук-
зиционирование связано с высокой транскрип-
леосомы. Обработка смесью нуклеаз может при-
водить к формированию участков ДНК, отлича-
ционной активностью и работой транскрипцион-
ных факторов [5]. Значимость позиционирова-
ющихся по длине от ожидаемых для нуклеосомы
ния нуклеосом проявляется во многих
(рис. 1в,д): часть нуклеосом будет обработана не-
биологических исследованиях и напрямую влия-
достаточно, а часть - чрезмерно. Таким образом,
ет на локальную структуру хроматина [6]. Пози-
сигналы в совокупности позволяют уверенно де-
ционирование нуклеосом может оказывать влия-
тектировать факт наличия нуклеосом на последо-
ние на процессы регуляции работы ДНК, напри-
вательности, однако их начальное позициониро-
мер, связывание нуклеосом с ДНК может
вание неоднозначно. Если считать центры сигна-
приводить к блокированию сайтов связывания
лов за диадные н.п. нуклеосом, то для каждого
других белков. Интересно, что с течением жизни
пика заселенности ожидается достаточно боль-
структура хроматина меняется: снижается уро-
шое число возможных положений нуклеосомы
вень экспрессии гистоновых белков, уменьшает-
(рис. 1г, линия 2, центры сигналов показаны чер-
ся заселенность генов нуклеосомами, что в ре-
ными точками). Таким образом, для точного
зультате приводит к нарушению процессов тран-
определения положений нуклеосом необходимо
скрипции [7].
дополнительно фильтровать и обрабатывать дан-
Как уже было отмечено выше, позициониро-
ные, как например в работах [11, 12].
вание нуклеосом зависит от последовательности
Эксперименты по определению позиций нук-
ДНК и активности хроматиновых ремоделлеров.
леосом по большей части получены для культур
Известно, что in vitro нуклеосомы способны к
клеток и содержат сигналы позиционирования
спонтанному тепловому передвижению. Однако
для большой совокупности геномов. На итоговых
механизм такого передвижения не до конца ясен,
профилях заселенности мы видим результат су-
в ряде работ по молекулярному моделированию
перпозиции множества альтернативных вариан-
были показаны начальные этапы перемещения
тов позиционирования, который дополнительно
нуклеосом по ДНК [8]. Один из основных мето-
смазан неоднородностью длин сигналов. Опреде-
дов изучения позиций нуклеосом in vivo - MNase
ление возможных расположений нуклеосом по
seq (от аббревиатуры МНКаза - микрококковая
таким данным - сложная вычислительная задача.
нуклеаза). В этом методе исследуют препарат хро-
В данной работе мы предлагаем метод отбора воз-
матина, выделенный из клеточной культуры. Та-
кой хроматин далее обрабатывают микрококко-
можных позиций нуклеосом по данным MNase-
вой нуклеазой - ферментом, разрезающим фраг-
секвенирования. Данный метод основан на отбо-
менты цепи ДНК, не связанные с белками.
ре наиболее вероятных положений нуклеосом по
Однако данный фермент предпочтительно разре-
изначальным сигналам с последующей фильтра-
зает AT-богатые регионы [9]. В результате после
цией допустимых комбинаций. Созданный нами
обработки получаются фрагменты ДНК, связан-
фильтр основан на геометрическом анализе до-
ные с белками (рис. 1в). В силу селективности эн-
ступного конформационного пространства для
донуклеазной активности и низкой экзонуклеаз-
молекулярных моделей нуклеосомных фибрилл.
ной активности, получившиеся фрагменты ДНК
Мы применяем данный метод для поиска доступ-
могут быть больше по размеру, чем связанные с
ных положений нуклеосом на ряде генов
белками участки. Чтобы уточнить области связы-
S. сerevisae по данным эксперимента MNase-се-
вания, образец дополнительно обрабатывают эк-
квенирования.
БИОФИЗИКА том 68
№ 5
2023
914
ВАСИЛЬЕВ и др.
Таблица 1. Характеристики исследуемых участков генома S. cerevisae
Число
Коэффициент
Число
Число
Открытая рамка
стерически
корреляции
Координата
Координата
возможных
возможных
считывания
возможных
модельного
начала ОРС
конца ОРС
цепочек
позиций
(ОРС)
позиций
профиля с
нуклеосом
нуклеосом
нуклеосом
экспериментом
YJR046W
522048
523912
154688
101
92
0.76
YOR066W
449436
451375
60843
101
93
0.49
YLR177W
511054
512990
84870
92
65
0.48
YFL041W
49139
51007
51543
89
80
0.51
YHL019C
67731
69548
22664
85
72
0.65
YPR155C
835563
837413
42262
94
82
0.46
МАТЕРИАЛЫ И МЕТОДЫ
как отступ от диадной н.п. В качестве шаблона
для создания нуклеосомных цепочек использова-
Обработка геномных данных и выбор областей
ли структуру 3LZ0 [17] (рис. 1а) из банка данных
для моделирования. Для разработки и апробиро-
PDB. Для этой структуры были рассчитаны пара-
вания метода использовали набор данных из ра-
метры ДНК и относительное положение белково-
боты [13]. Из базы архива секвенирований SRA
го ядра нуклеосомы при помощи программного
[14] был загружен набор чтений для эксперимента
пакета PyNAmod. Создание модели цепочки нук-
по MNase-секвенированию хроматина S. сerevis-
леосом происходило в несколько этапов: созда-
ae, идентификатор эксперимента SRR1802189.
вался набор параметров для ДНК требуемой дли-
Чтения были обработаны и картированы на ге-
ны в B-форме; в местах положений нуклеосом па-
ном дрожжей (версия сборки генома
раметры ДНК заменялись параметрами для
GCF_000146045.2) с помощью программы Bow-
нуклеосомы; проводилась конвертация парамет-
tie2 [15]. Полученные сигналы были отфильтро-
ров ДНК в реальное пространство; рассчитыва-
ваны по стандартному протоколу программы.
лись координаты центров нуклеосом.
Для дальнейшего анализа были выбраны отобра-
жения сигналов на (+) цепи ДНК. Для исследова-
Таким образом, данный подход позволяет со-
ния были выбраны шесть случайных генов дли-
единять нуклеосомы прямыми участками двухце-
ной менее 2000 н.п. При выборе участков для мо-
почечной ДНК для дальнейшего расчета их ха-
делирования использовали следующие критерии:
рактеристик. В частности, для цепочек нукле-
сигнал MNase-секвенирования определен от бо-
осом рассчитывали количество внутренних
лее чем 400 нуклеосом, распределение длин сиг-
стерических перекрываний. Для пар нуклеотидов
налов имеет максимум в 145 н.п., для данных ге-
был взят радиус 3.5 Å, а для белковых ядер нукле-
нов имеется аннотация уровнем экспрессии. Ан-
осом - 32 Å. Для каждой пары частиц рассчиты-
нотация уровнем экспрессии была взята из
вали перекрывание: если расстояние между ча-
эксперимента [16]. Список исследуемых генов и
стицами меньше суммы их заданных радиусов, то
их геномные координаты приведены в табл. 1. В
принимается, что частицы перекрываются, а со-
результате данной обработки были получены сиг-
ответствующая цепочка нуклеосом отвергается.
налы позиционирования нуклеосом внутри ис-
Для доступных конформаций нуклеосомных це-
следуемых генов.
почек рассчитывали радиус инерции по центрам
Моделирование цепочек нуклеосом. Для моде-
нуклеосом.
лирования цепочек нуклеосом использовали
Для проведения всех расчетов применяли про-
огрубленный геометрический подход. В таком
граммный пакет PyNAMod, для ускорения чис-
подходе каждая пара нуклеотидов и каждое бел-
ленной математики использовали библиотеки
ковое ядро нуклеосомы задается сферическими
Numpy и транслятор Numba.
частицами разных диаметров (рис. 1б). В данном
Для поиска доступных цепочек нуклеосом бы-
подходе описание геометрии фибриллы проводи-
ли созданы моделей всех возможных комбинаций
ли во внутренних параметрах ДНК: взаимные
цепочек из пяти нуклеосом и длиной соединяю-
ориентации н.п. относительно друга задавались
щей ДНК от 0 до 100 н.п. Всего было создано
при помощи шести параметров (shift, slide, rise,
tilt, roll, twist). Позиции нуклеосом записывали
108 комбинаций. Расчеты проводили в парал-
БИОФИЗИКА том 68
№ 5
2023
УТОЧНЕНИЕ ПОЗИЦИЙ НУКЛЕОСОМ ВНУТРИ ОТДЕЛЬНЫХ ГЕНОВ
915
лельном режиме с использованием 80 процессор-
(см. пункт
«Моделирование цепочек нукле-
ных ядер. Длина свободного участка ДНК на
осом»).
концах цепочек нуклеосом составляла 50 н.п. Це-
почки, в которых наблюдались стерические пере-
крывания, отбрасывались. Затем было рассчита-
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
но отношение числа доступных конформаций к
общему числу комбинационно возможных цепо-
Точное позиционирование важно с геометри-
чек нуклеосом. Для анализа цепочек с шестью и
ческой точки зрения, так как сдвиг нуклеосомы
семью нуклеосомами были рассчитаны конфигу-
на 1 н.п. приводит к небольшому смещению
рации с максимальной длиной линкерных обла-
(3.3 Å), но значительному повороту (34.3°). Вы-
стей 40, так как полный расчет на доступных
ровненные участки из секвенирования - сигналы
мощностях занял бы более трех лет.
позиционирования нуклеосом - не позволяют
Моделирование позиций нуклеосом в гене. Для
однозначно определить позицию диадных н.п.
предсказания возможных позиций нуклеосом в
нуклеосом. Обработка смесью нуклеаз приводит
гене из всех сигналов MNase секвенирования
к формированию участков ДНК, отличающихся
учитывали длину сигнала. В случае если длина
по длине от нуклеосомной (рис. 1в,д): часть нук-
фрагмента соответствовала длине нуклеосомной
леосом будет обработана недостаточно, а часть -
ДНК (145 пар нуклеотидов), предполагалось од-
чрезмерно. Число коротких и длинных сигналов
нозначное определение диадной н.п. Если фраг-
значительно меньше числа нормальных сигналов
мент отличался по длине от нуклеосомной ДНК,
(рис. 1д). Для того чтобы участок ДНК был атако-
предполагалось наличие двух возможных диад-
ван нуклеазой и стал короче, ДНК должна «от-
ных н.п. (рис. 1г). Более короткие сигналы могут
крутиться» от нуклеосомы. Вероятность одновре-
получаться по причине того, что фермент расще-
менного откручивания нуклеосомы с двух концов
пил часть нуклеосомной ДНК с одного из концов.
ниже, чем вероятность одностороннего откручи-
Аналогично два возможных положения диадной
вания, по этой причине для всех коротких сигна-
н.п. возникает в том случае, когда фрагмент длин-
лов мы предполагали две возможные позиции
нее 145 н.п. Из полученных возможных позиций
диадной н.п., соответствующие откручиванию
диадных н.п. рассчитывали профиль вероятности
ДНК либо с одной, либо с другой стороны. Ана-
обнаружения диадной н.п. по последовательно-
логично, мы предположили, что более длинные
сти гена. Согласно этому профилю, для каждого
участки ДНК содержат две возможные позиции.
сигнала выбиралась только одна позиция диад-
Из профилей встречаемости диадных н.п. для
ной н.п. В случае равной вероятности для центров
каждого сигнала отбирали наиболее распростра-
диад такой сигнал отбрасывался. Для последую-
ненную. Из рис. 1г видно, что если считать за по-
щего анализа были выбраны позиции диадных
зиции диадных н.п. центры сигналов (черные
н.п., которые были обнаружены в двух и более
точки), возникает большое количество дополни-
сигналах.
тельных вариантов позиционирования (линия 2),
По отобранным позициям диадных н.п. был
однако при использовании предположения, опи-
построен направленный граф, в вершинах кото-
санного выше, число возможных позиций значи-
рого находятся кандидаты в позиции нуклеосом,
тельно сокращается (линия 3). Такая процедура
а связи присваиваются для нуклеосом, находя-
отбора позиций нуклеосом позволяет сократить
щихся не ближе 145 н.п. и не дальше 215 н.п.
число возможных кандидатов в два-три раза (в за-
(среднее расстояние между нуклеосомами для
висимости от гена). В других работах для уточне-
S. cerevisae 165 н.п. [18]). Все возможные конфор-
ния позиций нуклеосом анализируют не изна-
мации фибрилл гена можно представить, как путь
чальные сигналы, а профили заселенности нук-
в этом графе. В качестве начальных вершин путей
леосом. Такие профили обычно обрабатывают
выбирались вершины, в которые не приходят
оконными фильтрами для локальной нормиров-
связи и они находятся на расстоянии не более 250
ки и сглаживания. На обработанном профиле за-
н.п. от начала гена. Аналогично, конечные вер-
селенности находят все пики, каждый из которых
шины - это такие вершины, из которых не начи-
рассматривают как предположительный центр
нается связь, а нуклеосомы находятся на расстоя-
нуклеосомной ДНК. Затем определяют кластеры
нии не более 250 н.п. от конца гена. Из всех на-
возможных пиков - группы таких пиков на рас-
чальных вершин находятся все возможные пути
стоянии менее 147 н.п. Далее при анализе выби-
до каждой конечной вершины. При построении
раются возможные комбинации позиций таким
пути применяли фильтр по допустимым комби-
образом, чтобы комбинации нуклеосом форми-
нациям длин линкерных областей: при добавле-
ровали максимально схожие с изначальными
нии каждой вершины в путь, если длина пути с
профили заселенности [11]. Однако такой подход
новой вершиной больше или равна пяти, прове-
основан на усреднении сигналов по множеству
ряется, что комбинация четырех последних длин
клеток и позволяет определять лишь средние по-
линкерных областей в пути стерически возможна
зиции нуклеосом в гене.
БИОФИЗИКА том 68
№ 5
2023
916
ВАСИЛЬЕВ и др.
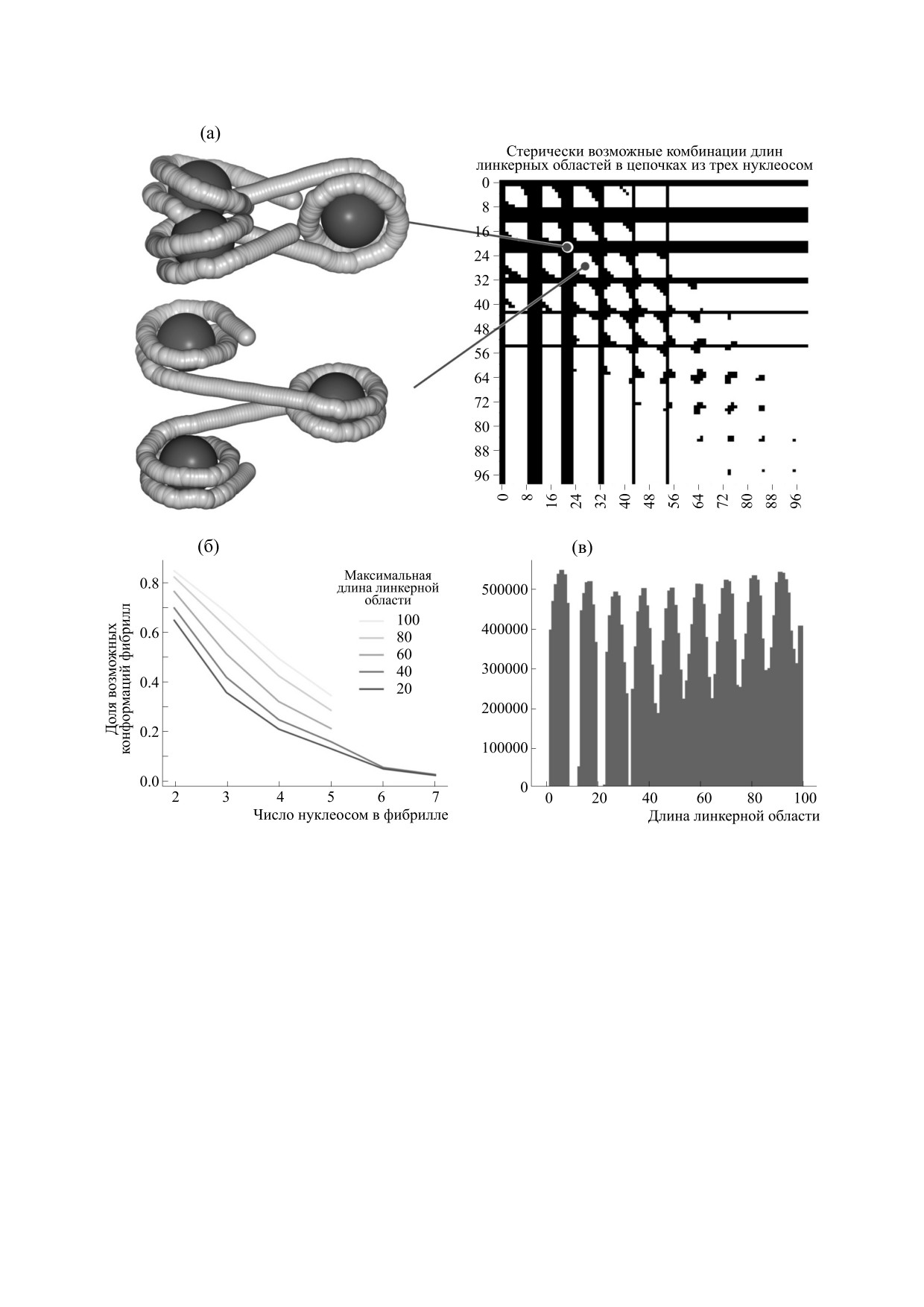
Рис. 2. (а) - Иллюстрация сканирования пространства доступных цепочек из трех нуклеосом. На тепловой карте черным
показаны заслоненные конфигурации, белым - разрешенные комбинации линкерных областей ДНК. Слева от карты
показаны примеры заслоненных и разрешенных цепочек. (б)
- График зависимости доли доступного
конфигурационного пространства от числа нуклеосом в цепочке. (в) - Распределение длин линкерных областей ДНК во
всех допустимых цепочках из пяти нуклеосом.
Геном S. cerevisae отличается сравнительно не-
комбинация позиций нуклеосом возможна гео-
большим размером и высокой долей содержания
метрически, нуклеосомы могут перекрываться
кодирующих последовательностей. В среднем на
(рис. 2а). Для того чтобы создать фильтр для отбо-
один ген у дрожжей приходится порядка 2000 н.п.
ра цепочек нуклеосом, мы рассчитали стериче-
Для генов со строгим позиционированием нукле-
ские перекрывания для цепочек длиной от двух
осом средняя длина линкерных областей ДНК
до пяти нуклеосом с длиной линкерных областей
составляет порядка 20 н.п. [18] Таким образом, на
ДНК от 0 до 100 н.п. и для цепочек длиной 7 и 8
один ген приходится порядка 10-11 нуклеосом.
для линкерных областей до 40 н.п. На рис. 2а вид-
Если позиция каждой нуклеосомы определена
но, что ряд комбинаций линкерных областей
неточно, возможное число комбинаций таких
приводит к появлению заслоненных структур, а
нуклеосом невероятно велико. Однако не любая
на тепловой карте видна периодичность. Такая
БИОФИЗИКА том 68
№ 5
2023
УТОЧНЕНИЕ ПОЗИЦИЙ НУКЛЕОСОМ ВНУТРИ ОТДЕЛЬНЫХ ГЕНОВ
917
периодичность хорошо согласуется с литератур-
сятся к соседним с ними позициям нуклеосом.
ными данными. Для регулярных нуклеосомных
Для всех доступных моделей был рассчитан ради-
фибрилл доступен ряд конфигураций, образую-
ус инерции от нуклеосомных частиц, который из-
щий две группы спиральных структур с характер-
менялся в пределах от 100 до 250 Å, а полученные
ными длинами линкерных областей 10N и 10N+5
модели значительно отличались геометрически
[19-21]. Для всех комбинаций фибрилл из 5 нук-
(рис. 3б,в). Мы не обнаружили зависимости меж-
леосом чаще всего встречаются ликеры длиной
ду определенными параметрами для нуклеосом-
10N+5 (рис. 2в). Интересно, что с ростом длины
ных цепочек (число доступных конформаций, ра-
нуклеосомных цепочек доля допустимых комби-
диус инерции) и уровнем экспрессии гена. Одна-
наций длин линкерных областей значительно па-
ко, так как эксперимент по определению уровня
дает, причем для малых длин линкерных областей
экспрессии и эксперимент по картированию нук-
она приближается к единицам процентов
леосом были проведены в разных условиях и на
(рис. 2б). Таким образом, для плотных цепочек
разных клеточных культурах, для них могут быть
нуклеосом доступно значительно меньшее кон-
характерны разные состояния хроматина.
фигурационное пространство чем для цепочек с
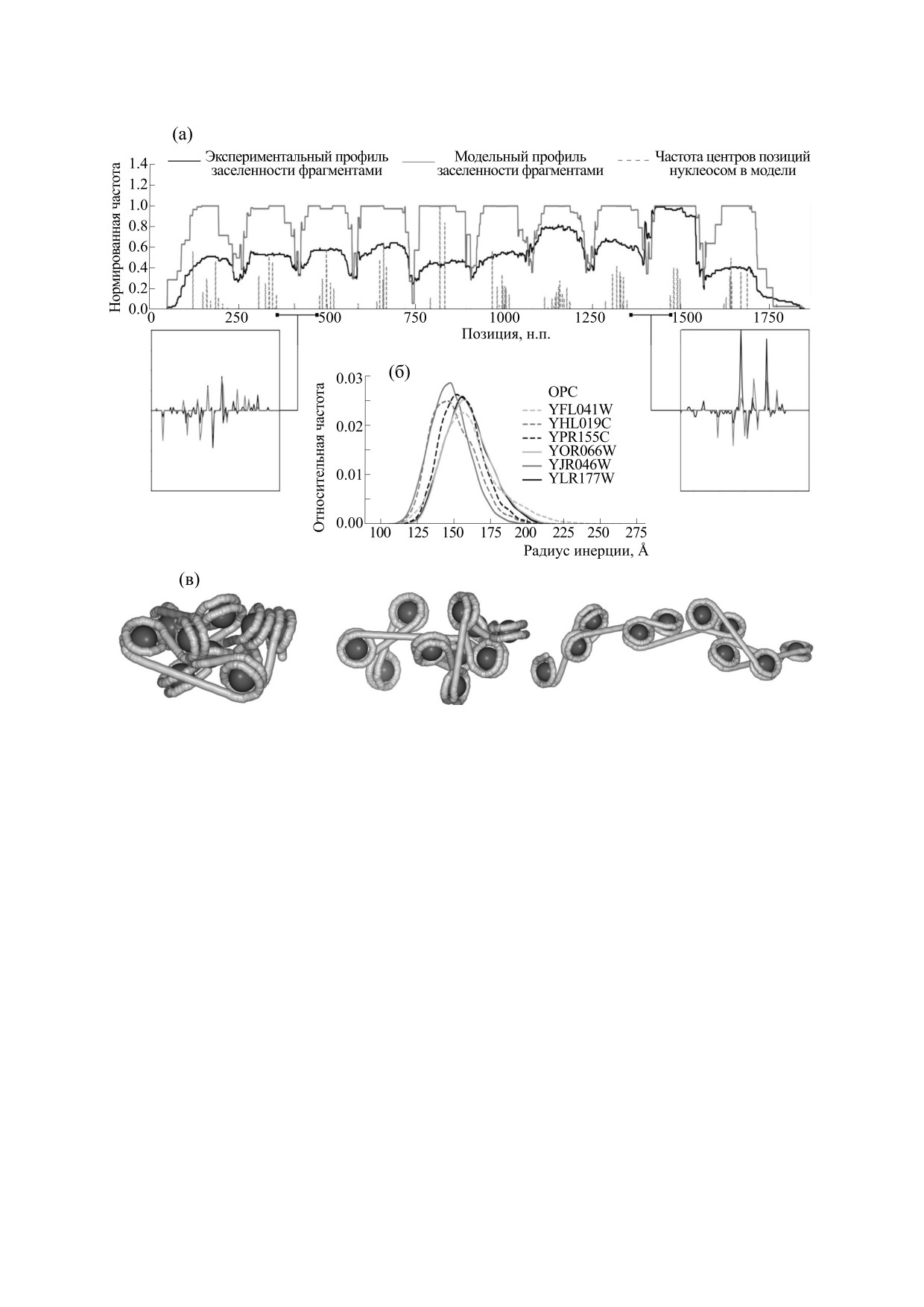
Интересно, что полученный модельный про-
большим межнуклеосомным расстоянием. Опи-
филь заселенности нуклеосом не только каче-
раясь на данное наблюдение, мы создали алго-
ственно совпадает с экспериментальным, но и
ритм отбора конфигураций нуклеосомных цепо-
схож с ним в деталях, о чем свидетельствует высо-
чек произвольной длины по экспериментальным
кий коэффициент корреляции Пирсона (рис. 3а,
данным.
коэффициенты корреляции представлены в
табл. 1). Таким образом, тонкая структура профи-
Мы провели поиск доступных конфигураций
лей в эксперименте является не шумом, а след-
цепочек нуклеосом для сигналов позициониро-
ствием наложения сигналов от нуклеосом из раз-
вания нуклеосом для шести генов S. cerevisae. Раз-
ных клеток. Следовательно, подходы, основан-
работанный нами подход основан на последова-
ные на сглаживании профилей заселенности
тельном переборе возможных комбинаций сиг-
нуклеосом с последующим поиском пиков, при-
налов нуклеосом на ДНК. Однако доступное
водят к потере сигнала точного позиционирова-
пространство комбинаций нуклеосом на гене
ния. Для профилей с ярко выраженными десятью
длиной 2000 н.п. слишком велико для прямого
пиками заселенности нуклеосом были обнаруже-
перебора. Так, после обработки сигналов MNase-
ны возможные цепочки из 9 и 11 нуклеосом. Доля
секвенирования для гена TAH11 (открытая рамка
таких цепочек сравнительно невелика (порядка
считывания YJR046W) мы отфильтровали
5%), однако данные цепочки содержат сигналы с
101 возможную позицию нуклеосомных н.п. Ис-
высокой интенсивностью из экспериментальных
ходя из карты заселенности сигналов (рис. 1е), на
данных. Интересно, что в модельных профилях
данном гене находится 10 нуклеосом. Для такого
заселенности нуклеосомами для всех генов (в от-
числа сигналов, число сочетаний из 10 нуклеосом
личие от экспериментальных) высоты пиков бы-
составляет порядка
1016 комбинаций, однако
ли равны между собой. Высота пика в экспери-
нуклеосомные сигналы не могут перекрываться.
менте не связана прямо с представленностью
Исходя из числа сигналов, для каждого пика засе-
нуклеосомы в геноме, а зависит от времени обра-
ленности нуклеосомами ожидается порядка
ботки ферментами и сродства нуклеаз к последо-
10 вариантов расположения нуклеосом, что экви-
вательности линкерных областей ДНК между
валентно 1010 возможных конфигураций цепочек
нуклеосомами [22].
Такое число позиций весьма затруднительно ис-
Разработанный нами подход содержит ряд
следовать прямым перебором. Для того чтобы не
ограничений. В данной модели мы не учитываем
рассчитывать заведомо недоступные конфигура-
гибкость ДНК. Такой подход может приводить к
ции нуклеосомных цепочек мы отбрасывали кон-
чрезмерной фильтрации конфигураций. Однако,
формацю, как только в ней встречалась запре-
учитывая длину персистентности ДНК порядка
щенная комбинация позиций для пяти нукле-
160 н.п. [23] и малую длину линкерных областей
осом. В итоге для гена TAH11 было обнаружено
ДНК в генах дрожжей, для реализации заслонен-
154688 возможных комбинаций позиций, что на
ных конфигураций потребуется значительно изо-
три порядка меньше изначальной оценки. Инте-
гнуть ДНК. Также при поиске возможных кон-
ресно, что в результате работы данного алгоритма
фигураций нуклеосомных цепочек, мы ограни-
девять возможных позиций нуклеосом не вошли
чивали максимальную длину линкерной области
ни в одну цепочку. Для остальных обработанных
70 н.п., что позволяет применять подход только
генов также была отфильтрована большая часть
для плотно заселенных нуклеосомами участков
нуклеосомных цепочек (табл. 1) и отсеяно от 8 до
хроматина.
27 возможных позиций нуклеосом. Все отсеян-
ные позиции встречались не более чем в двух сиг-
Разработанный нами алгоритм позволяет эф-
налах MNase-секвенирования и, вероятно, отно-
фективно отсеивать недоступные комбинации
БИОФИЗИКА том 68
№ 5
2023
918
ВАСИЛЬЕВ и др.
Рис. 3. (а) - Сравнение экспериментального и модельного профилей заселенности фрагментами нуклеосом. Показан
ген TAH11 (открытая рамка считывания YJR046W). На врезках показаны дифференциальные профили заселенности.
(б) - Распределение радиусов инерции модельных цепочек нуклеосом от разных генов пекарских дрожжей. (в) -
Визуализация фибрилл с минимальным, медианным и максимальным радиусами инерции.
нуклеосомных цепочек и конформационно за-
ций клеток, будут подавлены доминантными со-
прещенные экспериментальные сигналы. Одна-
стояниями.
ко после такой обработки в результате остаются
В данной работе предложен и апробирован
десятки и сотни тысяч доступных комбинаций
комбинированный метод отбора возможных нук-
нуклеосом. Большое число возможных вариантов
леосом по данным MNase-секвенирования и мо-
может быть объяснено тем, что в экспериментах
лекулярного моделирования. Разработанный ал-
по MNase-секвенированию наблюдается супер-
горитм позволяет отфильтровывать недоступные
комбинации нуклеосом и их позиции. Предло-
позиция нуклеосом из клеточной культуры. Кон-
женный алгоритм позволяет уточнять позиции
кретное количество геномов, подверженных ана-
нуклеосом на отдельных генах, однако его можно
лизу можно оценить исходя из оптической плот-
адаптировать для обработки сигнала от кластеров
ности культуры клеток и объема образца, в
нуклеосом на полном геноме.
эксперименте [13] анализу подвергалось порядка
108 клеток. Очевидно, что при таком количестве
клеток и случайном расположении нуклеосом в
БЛАГОДАPНОCТИ
каждой из них профили заселенности были бы
Работа выполнена с использованием оборудо-
равномерными. Учитывая, что на каждый ген
вания Центра коллективного пользования сверх-
приходится порядка 500 сигналов, состояния
высокопроизводительными вычислительными
хроматина, характерные для небольших популя-
ресурсами МГУ имени М.В. Ломоносова.
БИОФИЗИКА том 68
№ 5
2023
УТОЧНЕНИЕ ПОЗИЦИЙ НУКЛЕОСОМ ВНУТРИ ОТДЕЛЬНЫХ ГЕНОВ
919
ИCТОЧНИКИ ФИНАНCИPОВАНИЯ
6. D. S. Saxton and J. Rine, Proc. Natl. Acad. USA, 117
(44), 27493 (2020).
Работа выполнена при финансовой поддержке
7. J. Feser and J. Tyler, FEBS Lett., 585 (13), 2041 (2011).
Российского научного фонда (грант № 21-74-
8. G. A. Armeev, et al., Nature Commun., 12 (1), 2387
00033).
(2021).
9. C. Dingwall, G. P. Lomonossoff, and R. A. Laskey,
Nucl. Acids Res., 9 (12), 2659 (1981).
КОНФЛИКТ ИНТЕРЕСОВ
10. T.-H. S. Hsieh, et al., Cell, 162 (1), 108 (2015).
11. R. Schöpflin, et al., Bioinformatics, 29 (19), 2380
Авторы заявляют об отсутствии конфликта
(2013).
интересов.
12. X. Zhou, et al., eLife, 5, e16970 (2016).
13. H. A. Cole, et al., Nucl. Acids Res., 44 (2), 573 (2016).
СОБЛЮДЕНИЕ ЭТИЧЕСКИХ СТАНДАРТОВ
14. R. Leinonen, H. Sugawara, and M. Shumway, Nucl.
Acids Res., 39 (Database issue), D19 (2011).
Настоящая работа не содержит описания ка-
15. B. Langmead and S. L. Salzberg, Nature Methods, 9
ких-либо исследований с использованием людей
(4), 357 (2012).
и животных в качестве объектов.
16. K. Waern and M. Snyder, G3: Genes, Genomes, Ge-
netics, 3 (2), 343 (2013).
17. D. Vasudevan, E. Y. D. Chua, and C. A. Davey, J. Mol.
СПИСОК ЛИТЕРАТУРЫ
Biol., 403 (1), 1 (2010).
18. T. Tsukiyama, et al., Genes Dev., 13 (6), 686 (1999).
1. G. S. Omenn, Mol. Cell. Proteomics, 20, 100062
19. N. Kepper, et al., Biophys. J., 95 (8), 3692 (2008).
(2021).
20. D. Norouzi, et al., AIMS Biophys., 2 (4), 613 (2015).
2. P. T. Lowary and J. Widom, J. Mol. Biol., 276 (1), 19
21. V. B. Zhurkin and D. Norouzi, Biophys. J., 120 (4), 577
(1998).
(2021).
3. J. Ocampo, et al., Nucl/ Acids Res., 44 (10), 4625
22. R. V. Chereji, T. D. Bryson, and S. Henikoff, Genome
(2016).
Biol., 20 (1), 198 (2019).
4. G.-C. Yuan, et al., Science, 309 (5734), 626 (2005).
23. J. S. Mitchell, et al., J. Chem. Theory Comput., 13 (4),
5. W. Lee, et al., Nature Genet., 39 (10), 1235 (2007).
1539 (2017).
Updating Nucleosome Positions within Individual Genes Using Molecular Modeling
Methods and MNase Sequencing Data
V.A. Vasilev*, D.M. Ryabov*, A.K. Shaytan*, and G.A. Armeev*
Lomonosov Moscow State University, Leninskie Gory 1/12, Moscow, 119234 Russia
Organization of chromatin plays an important role in regulating the genetic machinery of the cell. The basic
unit of chromatin packaging is a nucleosome, which harbors DNA of about 145 base pairs in length. The
packaging of genetic material and its accessibility to transcription enzymes and other regulatory chromatin
proteins depends on the positions of nucleosomes. MNase sequencing is used to examine nucleosome posi-
tions in a genome. MNase sequencing data are sufficient for detecting the presence of nucleosomes on the
sequence, but a determination of the precise locations of nucleosomes can be problematic. Accurate deter-
mination of nucleosome positions requires additional data filtering and processing. In this study, using
MNase sequencing data, a combined method based on geometric analysis of nucleosome chain molecular
models is proposed for selecting possible nucleosome positions. The developed algorithm efficiently elimi-
nates inaccessible nucleosome chain combinations and conformationally prohibited nucleosome positions.
Keywords: chromatin, nucleosome, nucleosome positioning, molecular modeling
БИОФИЗИКА том 68
№ 5
2023