

Химия твердого топлива, 2021, № 3, стр. 3-11
АДСОРБЦИЯ ФЕНОЛА АКТИВИРОВАННЫМИ УГЛЯМИ НА ОСНОВЕ ИСКОПАЕМЫХ УГЛЕЙ РАЗНОЙ СТЕПЕНИ МЕТАМОРФИЗМА
Ю. В. Тамаркина 1, *, В. Н. Анищенко 1, **, А. Н. Редько 1, ***, В. А. Кучеренко 1, ****
1 Институт физико-органической химии и углехимии имени Л.М. Литвиненко НАН Украины
02160 Киев, Украина
* E-mail: Tamarkina@nas.gov.ua
** E-mail: Anischenko@nas.gov.ua
*** E-mail: A.M.Redko@nas.gov.ua
**** E-mail: V.O.Kucherenko@nas.gov.ua
Поступила в редакцию 01.10.2020
После доработки 05.11.2020
Принята к публикации 09.12.2020
Аннотация
Исследована адсорбция фенола активированными углями, полученными щелочным термолизом (КОН, 800°C) ископаемых углей разной степени метаморфизма (Cdaf = 70.4–95.6%). Получены кинетические зависимости и изотермы адсорбции в области начальных концентраций ≤ 3 мг/см3 (25°С). Определены константы скорости поглощения адсорбата, максимальные и удельные емкости по фенолу и их зависимости от Cdaf. Обсуждены основные процессы взаимодействия фенола с поверхностными адсорбционными центрами.
ВВЕДЕНИЕ
В настоящее время наблюдается высокий уровень загрязнения водных ресурсов различными органическими соединениями – экотоксикантами, такими как гербициды, пестициды, детергенты, красители. В эту группу также входят фенол и его производные, которые присутствуют в сточных водах предприятий фармацевтической, полимерной, резинотехнической и коксохимической промышленности [1, 2]. Фенольные соединения вредны для здоровья человека и при попадании в организм вызывают дегенерацию белка, эрозию тканей и повреждение внутренних органов [3]. Кроме того, при хлорировании загрязненной фенолами питьевой воды могут образовываться хлорфенолы – предшественники диоксинов, отнесенных к разряду наиболее высокотоксичных стойких органических загрязнителей, обладающих мутагенными и канцерогенными свойствами. Эти обстоятельства диктуют жесткую необходимость очистки воды от фенольных соединений.
Одними из наиболее эффективных методов улавливания фенола являются адсорбционные методы, которые используют различные материалы естественного и искусственного происхождения: ископаемые и активированные угли (АУ), шламы, слоистые глины, коксы, полимеры, ионообменные смолы, лигноцеллюлозные материалы [1]. Их адсорбционная емкость по фенолу (Аm) варьируется в широких пределах (Аm = 10–350 мг/г), но большие значения (Аm ≥ 100 мг/г) чаще всего характерны для АУ [1, 4]. Например, образцы АУ, полученные из биомассы сочетанием карбонизации (600–700°С, 1 ч) и активации гидроксидом калия (соотношение КОН/субстрат RKOH = 1 г/г, 850°С, 2 ч) имеют емкость Аm ≤ ≤ 140 мг/г [5] или Аm = 140–200 мг/г при повышенных значениях RKOH = 2–4 г/г (800°С, 1 ч) [6]. Карбонизаты (500–800°С, 5 ч) ионообменной смолы, насыщенной ионами калия, характеризуются значениями Аm ≤ 167 мг/г [7]. Образцы АУ из стирол-дивинилбензольного сополимера (активатор – СО2, 900°С) имеют емкость Аm ≤ 328 мг/г [8], а АУ из смесей целлюлозы и полистирола (активатор – Н2О, 800°С) – Аm = 312–417 мг/г [9]. Значения адсорбционной емкости того же порядка показывают буроугольные АУ (Аm = 240–320 мг/г), полученные щелочным термолизом (RKOH = 1–2 г/г, 800°С, 1 ч) [10], а также образцы АУ (Аm = 312–453 мг/г) из окисленных ископаемых углей с Cdaf = 63.0–84.6% (RKOH = 1 г/г, 800°С, 1 ч) [11]. Щелочная активация в таких же условиях каменных углей и антрацитов также приводит к образованию АУ с величиной удельной поверхности S ≤ 1500 м2/г [12] и относительно высокой адсорбционной емкостью по йоду ( ≤ 960 мг/г) и красителю метиленовому голубому ( ≤ 200 мг/г).
Цель данной работы – количественно оценить адсорбционную способность АУ по отношению к фенолу и установить влияние на этот параметр степени метаморфизма ископаемого угля.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В работе использованы ископаемые угли с возрастающим содержанием углерода, которое выбрано критерием степени метаморфизма (СМ), охватывающие диапазон Cdaf = 70.4–95.6% (табл. 1); подробная характеристика приведена в работе [12].
Таблица 1.
Объемы и удельная поверхность разных пор образцов АУ, полученных термопрограммируемой щелочной активацией углей разной степени метаморфизма
| Уголь | Активированный уголь | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| уголь | Сdaf, % | YАУ, % | объем пор, см3/г | удельная поверхность пор, м2/г | ||||||
| Vt | V1nm | Vmi | Vme + ma | S | S1nm | Smi | Sme + ma | |||
| БУ | 70.4 | 29.5 | 0.487 | 0.321 | 0.367 | 0.120 | 1142 | 1050 | 1120 | 22 |
| Д | 80.0 | 49.8 | 0.593 | 0.421 | 0.510 | 0.083 | 1547 | 1401 | 1535 | 22 |
| Г1 | 81.0 | 49.5 | 0.566 | 0.420 | 0.481 | 0.085 | 1488 | 1362 | 1468 | 20 |
| Г2 | 83.5 | 54.6 | 0.520 | 0.381 | 0.430 | 0.090 | 1345 | 1255 | 1323 | 22 |
| Ж | 85.0 | 55.1 | 0.564 | 0.395 | 0.489 | 0.075 | 1486 | 1366 | 1471 | 15 |
| К1 | 86.4 | 60.2 | 0.519 | 0.389 | 0.457 | 0.062 | 1354 | 1238 | 1343 | 11 |
| К2 | 88.6 | 60.8 | 0.495 | 0.380 | 0.427 | 0.068 | 1142 | 1054 | 1132 | 10 |
| ОС1 | 89.4 | 65.5 | 0.484 | 0.357 | 0.416 | 0.068 | 1196 | 1095 | 1188 | 8 |
| ОС3 | 90.8 | 70.3 | 0.448 | 0.340 | 0.393 | 0.055 | 1009 | 910 | 1001 | 8 |
| Т | 91.2 | 74.4 | 0.393 | 0.312 | 0.355 | 0.038 | 1083 | 1013 | 1076 | 7 |
| А | 93.3 | 74.5 | 0.307 | 0.176 | 0.251 | 0.056 | 681 | 511 | 659 | 22 |
| А5 | 95.6 | 82.8 | 0.229 | 0.036 | 0.169 | 0.059 | 322 | 77 | 305 | 17 |
Получение активированных углей (АУ) выполняли в условиях термопрограммируемой щелочной активации, включающей следующие стадии: 1) импрегнирование сухого угля водным раствором КОН (RKOH = 1.0 г/г) с последующей сушкой (120±10°С, ≥2 ч), 2) термопрограммируемое (4 град/мин) нагревание образца (~40 г) в атмосфере аргона до 800°С и выдержка 1 ч, 3) охлаждение, отмывка от КОН, сушка. Выход АУ обозначен как YАУ (ошибка измерения ±1%). Полученные образцы обозначены как АУ(Х), где Х-марка угля (табл. 1).
Характеристики пористой структуры АУ определены на основании изотерм низкотемпературной (77 K) адсорбции–десорбции азота (прибор Micromeritics ASAP 2020) после дегазации АУ (20 ч, 200°С). Общий объем пор Vt (см3/г) определяли по количеству N2, адсорбированного при относительном давлении p/p0 ~1.0. Объемы микропор (Vmi) и субнанопор (V1nm) определяли из интегральных зависимостей объема пор от средней ширины пор (W, нм), полученных методом 2D-NLDFT [13]. Суммарный объем мезо- и макропор (Vme+ma) оценивали по разности Vt – Vmi. Величину удельной поверхности АУ (S, м2/г) и поверхности субнано- (S1nm) и микропор (Smi) определяли из интегральных зависимостей S от W. Также рассчитывали доли субнанопор (V1nm/Vt), микропор (Vmi /Vt) и сумму долей макро- и мезопор (Vmе+mа/Vt).
Адсорбционную емкость по фенолу (А, мг/г) определяли аналогично методике [9]. Навеску высушенного при 120 ± 10°С образца АУ (0.200 г) помещали в колбу Эрленмейера, вводили раствор фенола (100 см3) заданной начальной концентрации (С0 = 0.001–3.0 мг/см3) и встряхивали при 25°С. По истечении заданного времени смесь фильтровали и измеряли оптическую плотность раствора с помощью спектрофотометра Perkin-Elmer Lambda 20 при длине волны 270 нм. Концентрацию фенола определяли сравнением с калибровочным графиком. Адсорбционную емкость рассчитывали по формуле А = (С0 – С) ⋅ V/m, где С0 и С – начальная и конечная концентрации адсорбата, V – объем раствора (100 см3), m – навеска АУ. Конечная концентрация С является текущей концентрацией Сτ при заданном времени τ (изучение кинетики адсорбции) или равновесной концентрацией Се (при регистрации изотерм адсорбции). Для каждого образца АУ определяли максимальную адсорбционную емкость Аm = Ае при С0 = = 3.0 мг/дм3. Изотермы адсорбции фиксировали без добавления какого-либо буферного раствора для стабилизации pH, чтобы избежать присутствия нового электролита в системе “АУ-фенол-Н2О”. Также рассчитывали удельную адсорбционную емкость АS = Аm/S (мг/м2), которая пропорциональна концентрации адсорбционных центров (АЦ) на поверхности АУ. Дополнительно рассчитывали степень заполнения (coverage factor [14]) поверхности АУ молекулами фенола CF = = AS · 10–3 ⋅ М–1 ⋅ NA ⋅ SФ, где М – молекулярная масса фенола (94.11 г/моль), NA – число Авогадро, SФ – площадь молекулы фенола. Для этих расчетов постулировано, что молекулы фенола укладываются плотно друг к другу и параллельно поверхности АУ. Размеры молекулы фенола 0.58 нм × 0.43 нм [14].
Даные по кинетике адсорбции фенола аппроксимировали с использованием моделей псевдо-первого (1) и псевдо-второго (2) порядков, которые часто применяют для описания адсорбционных процессов в неравновесных условиях. Для оценки влияния транспортных эффектов использовали модель (3) внутричастичной диффузии [5, 11, 15]:
(1)
$A = {{A}_{e}}[{\text{1}} - {\text{exp}}\left( { - {{k}_{{\text{1}}}}{{\tau }}} \right)]\quad ({\text{I - модель),}}$(2)
$A = {{k}_{{\text{2}}}}A_{r}^{{\text{2}}}{{\tau /(1}} + {{k}_{{\text{2}}}}{{A}_{e}}{{\tau )}}\quad {\text{(II - модель),}}$(3)
$A = {{k}_{d}}{{{{\tau }}}^{{{\text{0}}{\text{.5}}}}} + C\quad \left( {{\text{ВД - модель}}} \right),$Для расчета изотерм адсорбции использовали модели Ленгмюра (4), Фрейндлиха (5), Темкина–Пыжова (6) и Редлиха–Петерсона (7) [5, 14, 16]:
(4)
${{A}_{e}} = {{A}_{L}}{{k}_{L}}{{C}_{e}}{\text{/1}} + {{k}_{L}}{{C}_{e}}\quad (L{\text{ - модель}}),$(5)
${{A}_{e}} = {{k}_{{\text{F}}}} \cdot {\text{C}}_{e}^{{1/{{n}_{{\text{F}}}}}}\quad (F{\text{ - модель}}),$(6)
${{A}_{e}} = {{B}_{{TP}}} \cdot \ln {{k}_{{TP}}} + {{B}_{{TP}}} \cdot \ln {{C}_{e}}\quad (TP{\text{ - модель}}),$(7)
${{A}_{e}} = {{A}_{{RP}}}{{C}_{e}}{\text{/}}({\text{1}} + {{B}_{{RP}}}C_{e}^{g})\quad (RP{\text{ - модель}}),$Аппроксимацию изотерм адсорбции выполняли минимизацией нормированного среднеквадратичного отклонения [17]:
где N – число экспериментальных точек, Ае(э) и Ае(р) – экспериментальное и расчетное значение равновесной адсорбционной емкости соответственно. Аналогично аппроксимировали кинетические кривые адсорбции – определяли минимум величины ∆А (%). Применимость моделей для описания адсорбции в системе “АУ-фенол-Н2О” оценивали по величине коэффициента детерминации (R2).ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Выход твердого продукта и характеристики пористой структуры АУ, полученных в одинаковых условиях щелочной активации, существенно зависят от СМ исходного угля (табл. 1). В ряду от бурого угля (БУ) до антрацита (А5) выход АУ линейно возрастает в соответствии с корреляционным уравнением YАУ = 2.00 · Сdaf – 112.3 (R2 = = 0.975).
В том же ряду общий объем пор Vt увеличивается при переходе от АУ(БУ) к АУ(Д), а затем уменьшается до минимальной величины у АУ(А5). Близкая тенденция прослеживается и для параметра Vmi: адсорбенты с наибольшей микропористостью образуются из углей с Cdaf = 80.0–86.4%. В ряду АУ из углей разной СМ доля микропор Vmi/Vt увеличивается почти линейно с 75.4% у АУ(БУ) до 90.3% у АУ(Т), а при переходе к АУ(А5) снижается до 73.8%, но остается доминирующей. То есть все полученные АУ являются микропористыми материалами.
Субнанопоры с W ≤ 1 нм присутствуют во всех АУ, но в антрацитовых АУ их объем минимален. Доля этих пор в общем объеме пор АУ(А5) составляет 15.7%, у АУ(А) – 57.3%, тогда как у остальных АУ варьируется в интервале 65.9–79.4%. Суммарный объем мезо- и макропор Vme+ma с ростом Cdaf проявляет тенденцию к линейному снижению, но коэффициент детерминации невелик (R2 = 0.812). Удельная поверхность S максимальна у образцов АУ, полученных из каменных углей с Сdaf = 80.0–86.4%. Значения S уменьшаются при переходе к буроугольным и антрацитовым АУ (табл. 1).
Доминирующий вклад в величину S образцов АУ вносит поверхность микропор Smi: ее доля Smi/S варьируется в диапазоне 94.7–99.4%. Для АУ из бурого и каменных углей также высока доля субнанопор, которая составляет 90.2–93.5%, но снижается до 23.9% у АУ(А5). Поверхность мезо- и макропор мала (табл. 1), так что адсорбционные свойства полученных АУ будут определяться свойствами их микропористой структуры.
Кинетические зависимости поглощения фенола и изотермы адсорбции получены для образцов АУ(БУ), АУ(Д), АУ(Т) и АУ(А5). С течением времени адсорбционная емкость по фенолу возрастает с выходом на плато (рис. 1, 2). Для буроугольного АУ(БУ) равновесное значение Ае = = 240 мг/г достигается за 1.5–2.0 ч, для остальных образцов – примерно за 3 ч и составляет: Ае = = 293 мг/г для АУ(Д), Ае = 243 мг/г для АУ(Т) и Ае = 141 мг/г для АУ(А5). Кинетические данные аппроксимированы моделями I и II (уравнения 1 и 2) при условии Ае(э) = Ае(р). Параметры кинетических моделей приведены в табл. 2.
Рис. 1.
Кинетика адсорбции фенола образцами АУ(БУ) и АУ(Т): пунктирная линия – модель I, сплошная линия– модель II.
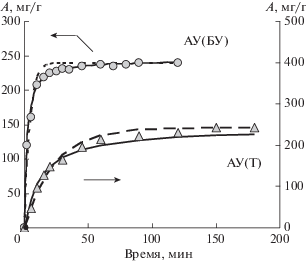
Рис. 2.
Кинетика адсорбции фенола образцами АУ(Д) и АУ(А5): пунктирная линия – модель I, сплошная линия– модель II.

Таблица 2.
Параметры кинетических моделей псевдо-первого и псевдо-второго порядка для адсорбции фенола образцами АУ из углей БУ, Д, Т и А5
| Кинетическая модель | Параметр | АУ(БУ) | АУ(Д) | АУ(Т) | АУ(А5) |
|---|---|---|---|---|---|
| Порядок I | Ае(э), мг/г | 240 | 293 | 243 | 139 |
| k1 · 10–2, мин–1 | 24.7 | 9.4 | 4.3 | 2.4 | |
| h0, мг г–1 мин–1 | 59.4 | 27.6 | 10.5 | 3.4 | |
| ∆А, % | 7.4 | 5.0 | 6.8 | 9.0 | |
| R2 | 0.970 | 0.985 | 0.984 | 0.994 | |
| Порядок II | k2 · 10–4, г мг–1 мин–1 | 21.7 | 6.47 | 3.20 | 2.66 |
| h0, мг г–1 мин–1 | 125.3 | 55.6 | 18.8 | 5.3 | |
| ∆А, % | 3.0 | 6.3 | 14.3 | 18.3 | |
| R2 | 0.993 | 0.988 | 0.977 | 0.953 |
Судя по величинам коэффициентов R2 (табл. 2), в ряду образцов от АУ(БУ) до АУ(А5) аппроксимация моделью псевдо-первого порядка становится точнее, а модель псевдо-второго порядка становится менее корректной. В том же ряду константа скорости k1 уменьшается примерно в 10 раз, а константа k2 – в 8 раз. Начальные скорости поглощения фенола снижаются еще разительнее: в 17.5 раза для модели I и в 23.6 раза для модели II, т.е. увеличение степени метаморфизма исходного угля как предшественника АУ приводит к существенному снижению скорости поглощения фенола (рис. 3).

Обращает на себя внимание независимость кинетических параметров от величины удельной поверхности АУ из углей БУ, Д, Т и А5. При переходе от АУ(БУ) к АУ(Д) поверхность увеличивается в 1.35 раза – с 1142 м2/г до 1547 м2/г (табл. 1), а начальные скорости адсорбции фенола снижаются в 2.2 раза; константа скорости k1 уменьшается в 2.6 раза, k2 – в 3.4 раза. Образцы АУ(БУ) и АУ(Т) имеют практически одинаковую поверхность (1142 и 1083 м2/г) (табл. 1), а константы скорости ниже в 5.7 раза (k1 и h0 в модели I) и 6.7 раза (k2 и h0 в модели II). Очевидно, что в условиях щелочной активации образуются АУ с разными по активности поверхностными адсорбционными центрами (АЦ), скорость взаимодействия которых с фенолом монотонно снижается с ростом СМ исходного угля (рис. 3).
Приведенные кинетические модели не позволяют оценить вклад диффузии в процесс адсорбции фенола. Для выявления транспортных эффектов применена диффузионная модель (уравнение 3) в графическом варианте, использованном в работе [5, 6, 11]. Если скорость определяющей стадией является диффузия адсорбата внутрь частиц АУ, то зависимость адсорбционной емкости А от τ1/2 передается прямой линией, проходящей через начало координат. Если скорость адсорбции лимитируется диффузией фенола через пограничный адсорбционный слой, то в уравнении (3) появляется отсекаемый отрезок С. Наличие нескольких линейных участков свидетельствует о присутствии разных механизмов адсорбции.
В нашем случае зависимости А от τ1/2 для всех АУ показывают два линейных участка (рис. 4). Первые участки можно отнести к диффузии фенола в мезопоры с константами kd1, значения которых проявляют тенденцию снижаться с увеличением СМ исходного угля (табл. 3). Для образцов АУ(Д) и АУ(Т) линии первых участков зависимостей А от τ1/2 проходят близко к началу координат, величины отсекаемых отрезков Cd1 малы и можно заключить, что скорость адсорбции фенола этими АУ лимитируется внутричастичной диффузией. Величина Cd1 при адсорбции на антрацитовом АУ(А5) выше (табл. 3), вероятно, вследствие затрудненности диффузии фенола через адсорбционный слой на поверхности АУ. Скорость адсорбции на буроугольном АУ(БУ) в начальный период явно лимитируется диффузией фенола через пограничный адсорбционный слой (Cd1 = 66.1 мг/г).

Таблица 3.
Параметры модели внутричастичной диффузии фенола в образцы АУ из ископаемых углей БУ, Д, Т и А5
| Образец | Параметр | ||||||
|---|---|---|---|---|---|---|---|
| kd1, мг г–1 мин–0.5 | kd2, мг г–1 мин–0.5 | kd1/kd2 | Cd1, мг г–1 | Cd2, мг г–1 | $R_{{d1}}^{2}$ | $R_{{d2}}^{2}$ | |
| АУ(БУ) | 41.4 | 2.62 | 15.8 | 66.1 | 214.9 | 0.964 | 0.893 |
| АУ(Д) | 52.6 | 3.85 | 13.7 | 11.8 | 244.4 | 0.957 | 0.897 |
| АУ(Т) | 31.7 | 5.55 | 5.7 | –6.7 | 170.9 | 0.948 | 0.960 |
| АУ(А5) | 16.5 | 6.17 | 2.7 | 20.5 | 58.2 | 0.971 | 0.999 |
Вторые линейные участки можно отнести к диффузии в микропоры и далее в субнанопоры. Здесь величины констант kd2 существенно ниже (табл. 3), причем в ряду образцов от АУ(БУ) к АУ(А5) значения kd2 растут, а соотношение констант kd1/kd2 убывает с 15.8 до 2.7. Видно, что с ростом СМ исходного угля (от бурого угля до антрацита) кинетические характеристики адсорбции фенола в начальный период и на заключительных стадиях заметно сближаются, т.е. для антрацитовых АУ процесс поглощения фенола становится более одинаковым для любого этапа адсорбции.
Для образцов АУ(БУ), АУ(Д) и АУ(Т) получены изотермы адсорбции фенола в диапазоне равновесных концентраций с0 ≤ 2.4 мг/см3 (рис. 4).
L-модель постулирует, что поверхность АУ химически однородна и максимальная адсорбционная емкость соответствует насыщенному монослою адсорбата. В ряде работ [5, 6, 9, 11, 14] она наиболее подходит для описания поглощения фенола и в нашем случае хорошо описывает изотермы образцов АУ(Д) и АУ(Т) из каменных углей, но расчетные величины емкости монослоя AL выше (на ~21%) экспериментальных значений Ае (табл. 4). Для буроугольного АУ эта модель пригодна плохо (R2 ≤ 0.921), хотя значения AL и Ае близки.
Таблица 4.
Параметры моделей изотерм адсорбции фенола образцами АУ(БУ), АУ(Д) и АУ(Т)
| Модель изотермы адсорбции | Параметр | Образец активированного угля | ||
|---|---|---|---|---|
| АУ(БУ) | АУ(Д) | АУ(Т) | ||
| L-модель | AL, мг г–1 | 230.7 | 353.7 | 293.8 |
| kL ⋅ 103, дм3 мг–1 | 10.2 | 2.23 | 2.14 | |
| ∆Ae, % | 46.4 | 3.2 | 2.0 | |
| R2 | 0.921 | 0.997 | 0.997 | |
| F-модель | kF, мг г–1 | 33.04 | 16.86 | 13.24 |
| nF | 3.75 | 2.64 | 2.59 | |
| ∆Ae, % | 18.2 | 10.0 | 11.4 | |
| R2 | 0.983 | 0.971 | 0.969 | |
| TP-модель | kTP, дм3 мг–1 | 1.164 | 0.023 | 0.021 |
| B | 28.70 | 76.61 | 64.52 | |
| ∆Ae, % | 28.7 | 3.3 | 2.0 | |
| R2 | 0.967 | 0.997 | 0.997 | |
| RP-модель | ARP, дм3 г–1 | 40.505 | 0.879 | 0.700 |
| BRP ⋅ 103, (дм3 мг–1)–g | 970 | 3.91 | 3.77 | |
| g | 0.7670 | 0.9425 | 0.9416 | |
| ∆Ae, % | 6.5 | 2.4 | 1.6 | |
| R2 | 0.990 | 0.998 | 0.998 | |
F-модель предполагает многослойную адсорбцию на химически неоднородной поверхности АУ, содержащей разные по активности адсорбционные центры (АЦ). Фактор неоднородности nF указывает на химическую (nF < 1) или физическую (nF > 1) адсорбцию [16]. Эта модель лучше описывает изотерму адсорбции образцом АУ(БУ), но для АУ(Д) и АУ(Т) дает наименьшие коэффициенты детерминации. Полученные значения nF = 2.59–3.75 (табл. 4) указывают на физическую адсорбцию фенола.
TP-модель учитывает эффект взаимодействия адсорбат/адсорбат и постулирует, что тепло адсорбции молекул в монослое линейно уменьшается с увеличением степени покрытия поверхности, а энергия связи характеризуется равномерным распределением до некоторого максимального значения [16]. Она хорошо описывает изотермы адсорбции фенола образцами АУ(Д) и АУ(Т) (R2 = = 0.997) и показывает близкие значения констант kTP и В (табл. 4). Для АУ(БУ) величина kTP значительно больше, константа В – в 2.2–2.7 раза ниже и данная модель хуже описывает изотерму по сравнению с моделью Фрейндлиха.
RP-модель объединяет черты L- и F-моделей, т.е. предполагает отклонения от идеальной модели мономолекулярной адсорбции. На это указывает отклонение коэффициента g от единицы (0 < g < 1), а при g = 1 эта модель трансформируется в модель Ленгмюра. RP-модель описывает изотермы наших АУ наилучшим образом (R2 ≥ 0.990, среднеквадратичное отклонение ∆Ae ≤ 6.5%), что передается линиями на рис. 4.
Следует отметить, что для изотермы образца АУ(БУ) из бурого угля все четыре модели дают значения коэффициентов уравнений (4)–(8), которые сильно отличаются от коэффициентов для АУ из каменных углей. Нормированное среднеквадратичное отклонение также велико и варьируется в пределах ∆Ae = 6.5–46.4%. Изотерма адсорбции фенола образцом АУ(БУ) визуально выглядит как наложение двух изотерм с изломом при равновесной концентрации Се = 0.5–0.6 мг/см3. Изотермы такого вида относят к типу L4, а их появление объясняют усилением поглощения при более высоких концентрациях фенола за счет многослойной адсорбции, пространственной переориентацией адсорбированных молекул из плоского положения в вертикальное и адсорбцией на разных по природе адсорбционных центрах (АЦ) [9]. В целом полученные данные свидетельствуют о значительной структурной неоднородности поверхности АУ из ископаемых углей.
Для всех АУ определены максимальные адсорбционные емкости по фенолу Am (табл. 5). С ростом СМ исходного угля значение Am увеличивается до максимального Am = 293 мг/г у АУ(Д), а затем снижается к антрацитам (рис. 5, линия 1). Удельная емкость АS, характеризующая адсорбционную способность 1 м2 поверхности АУ, мало меняется с увеличением Cdaf, варьируется в диапазоне АS = 0.189–0.438 мг/м2 и заметно увеличивается при переходе к антрацитовым АУ (рис. 6, линия 2). Видим, что в принятых условиях щелочной активации группа каменных углей образует АУ с достаточно близкими характеристиками пористой структуры (табл. 1) и адсорбционными свойствами (табл. 5). Наиболее метаморфизованный уголь – антрацит А5 трансформируется в АУ с меньшей удельной поверхностью и, как следствие, минимальной емкостью по фенолу. В то же время, судя по более высоким величинам удельной емкости АS (табл. 5), число АЦ на поверхности антрацитового АУ в 1.5–2.0 раза выше, чем у остальных образцов. Это обеспечивает более высокую степень заполнения поверхности при адсорбции фенола (CF ≈ 70%). Поскольку каркас антрацита состоит из пространственно агрегированных полиареновых структур (графеновых фрагментов), можно предположить, что антрацитовый АУ наследует это свойство исходного материала. В этом случае возможно более сильное электронодонорно-акцепторное взаимодействие фенола с π-системой графена [18], что может быть причиной повышенных значений удельной адсорбционной емкости АУ(А) и АУ(А5).
Таблица 5.
Адсорбционные свойства АУ из угля разной степени метаморфизма
| Образец АУ | Параметр | |||
|---|---|---|---|---|
| S, м2/г | Аm, мг/г | АS, мг/м2 | CF, % | |
| АУ(БУ) | 1142 | 240 | 0.210 | 33.54 |
| АУ(Д) | 1547 | 293 | 0.189 | 30.23 |
| АУ(Г1) | 1488 | 289 | 0.194 | 31.00 |
| АУ(Г2) | 1345 | 268 | 0.199 | 31.80 |
| АУ(Ж) | 1486 | 277 | 0.186 | 29.75 |
| АУ(К1) | 1354 | 261 | 0.193 | 30.77 |
| АУ(К2) | 1142 | 240 | 0.210 | 33.54 |
| АУ(ОС1) | 1196 | 261 | 0.218 | 34.83 |
| АУ(ОС3) | 1009 | 235 | 0.233 | 37.17 |
| АУ(Т) | 1083 | 243 | 0.224 | 35.81 |
| АУ(А) | 681 | 203 | 0.298 | 47.58 |
| АУ(А5) | 322 | 141 | 0.438 | 69.89 |
Рис. 5.
Изотермы адсорбции фенола образцами АУ(БУ) (1), АУ(Д) (2) и АУ(Т). Сплошные линии – изотермы Редлиха–Петерсона (RP-модель).

Рис. 6.
Зависимости адсорбционной емкости по фенолу Аm (1) и AS (2) активированных углей от содержания углерода исходного угля.

Если принять во внимание только величину удельной поверхности АУ (табл. 1), то обнаруживается следующее. С ростом S значения Аm линейно растут (рис. 6, линия 1) и аппроксимируются уравнением Аm = 0.115 · S + 113.71 (R2 = 0.971). В том же ряду образцов АУ удельная емкость по фенолу АS снижается (рис. 6, линия 2) и удовлетворительно аппроксимируется степенной зависимостью АS = 10.779 · S–0.5543 (R2 = 0.990). Характер зависимостей параметров Аm и АS от удельной поверхности микропор Smi и субнанопор S1nm идентичен приведенным на рис. 6. Соответствующие корреляционные уравнения следующие: для микропор Аm = 0.115 · Smi + 115.63 (R2 = 0.971) и АS = = 9.581 · (Smi)–0.5387 (R2 = 0.990), для субнанопор – Аm = 0.106 · S1nm + 137.27 (R2 = 0.971) и АS = 1.668 · · (S1nm)–0.296 (R2 = 0.954). Судя по близости коэффициентов зависимостей Аm от S, Smi и S1nm и их линейности, фенол одинаково адсорбируется на поверхности пор любого размера.
Рис. 7.
Зависимости адсорбционной емкости по фенолу Ае (1) и AS (2) от величины удельной поверхности АУ.

Также необходимо отметить следующее. Исследованный в данной работе бурый уголь отличается от остальных углей генетически, поскольку сформирован из иного палеорастительного материала. Если образец АУ(БУ) исключить из рассмотрения, то для АУ из каменных углей (диапазон Cdaf = 80.0–95.6%) прослеживается следующая общая закономерность. Увеличение СМ исходного угля уменьшает пористость и удельную поверхность АУ, снижает максимальную емкость по фенолу, увеличивает концентрацию поверхностных АЦ (растет удельная емкость), но ослабляет взаимодействие АЦ с фенолом и, как следствие, уменьшает константы скорости адсорбции.
При обсуждении механизма адсорбции фенола на углеродных адсорбентах основными считают следующие три процеса: 1) π-π-электронное взаимодействие, 2) формирование электроно-донорно-акцепторных (ЭДА) комплексов, 3) образование водородных связей [1, 4, 9].
Первый определяется взаимодействием π-электронов фенольного кольца и π-электронов графенового слоя в АУ, которое усиливается при адсорбции в субнанопорах за счет увеличенного адсорбционного потенциала соседних графеновых стенок. Второй процесс – образование ЭДА-комплексов между поверхностными электронодонорными группами (например, карбонилами) и ароматическим кольцом фенола, действующим как электроноакцептор [9]. Формирование ЭДА-комплексов требует определенного “стерического простора” и реализуется в микропорах и мезопорах. Третий процесс обусловлен образованием водородных связей ОН-групп фенола с поверхностными функциональными группами. Можно предположить, что адсорбция фенола на полученных АУ включает все вышеперечисленные процессы, но их вклад меняется с ростом степени метаморфизма исходного ископаемого угля. При адсорбции фенола в субнанопорах доминирующим процессом можно считать π-π-взаимодействие с графенами, которые пространственно расположены на расстояниях менее 1 нм друг от друга и жестко связаны одинарными ариленовыми Сar–Car–связями [12].
ВЫВОДЫ
1. Степень метаморфизма (СМ) ископаемого угля, определяемая содержанием углерода (Cdaf), существенно влияет на характеристики пористой структуры и адсорбционные свойства активированных углей (АУ), полученных термолизом с КОН (800°С) при невысоком соотношении КОН/уголь (1 г/г).
2. С ростом Cdaf в диапазоне 70.4–95.6% максимальная адсорбционная емкость по фенолу увеличивается от 240 мг/г до максимальной 293 мг/г (АУ из длиннопламенного угля с Cdaf = 80.0%) и снижается к антрацитовым АУ до 141 мг/г.
3. Кинетика адсорбции фенола при 25°С подчиняется моделям псевдо-первого и псевдо-второго порядков, но с ростом СМ угля точность первой улучшается (рост коэффициента R2 c 0.970 до 0.994), а применение второй модели становится менее корректным (снижение R2 c 0.993 до 0.953). Увеличение СМ угля как предшественника АУ снижает скорость адсорбции, что выражается в уменьшении констант скорости поглощения фенола: k1 – в 10 раз, k2 – в 8 раз. Изотермы адсорбции фенола наилучшим образом описываются моделью Редлиха–Петерсона (R2 ≥ 0.990).
4. С ростом удельной поверхности АУ значения максимальной адсорбционной емкости по фенолу линейно возрастают и описываются корреляционным уравнением Аm = 0.115 · S + 113.71 (R2 = 0.971). В том же ряду удельная емкость, пропорциональная концентрации поверхностных адсорбционных центров, снижается в соответствии со степенной зависимостью АS = 10.779 · S–0.5543 (R2= 0.990).
5. Для АУ из каменных углей и антрацитов прослеживается общая закономерность с ростом СМ – снижение пористости и удельной поверхности АУ, уменьшение величины емкости по фенолу, но увеличение удельной емкости, пропорциональной концентрации поверхностных адсорбционных центров.
Список литературы
Lin S.-H., Juang R.-S. // J. Environm. Management. 2009. V. 90. № 3. P. 1336. https://doi.org/10.1016/j.jenvman.2008.09.003
Tran V.S., Ngo H.H., Guo W., Zhang J., Liang S., Ton-That C., Zhang X. // Bioresource Technology. 2015. V. 182. P. 353. https://doi.org/10.1016/j.biortech.2015.02.003
Вредные вещества в промышленности. Справочник для химиков, инженеров и врачей. Изд. 7-е, пер. и доп. Т. 1. Органические вещества. Под ред. Н.В. Лазарева и Э.Н. Левиной. Л.: Химия. 1976. 592 с.
Bansal R.C., Goyal M. Activated carbon adsoption. Tayler&Francis Group: Boca Raton. 2005. 472 p.
Hameed B.H., Rahman A.A. // J. Hazard. Materials. 2008. V. 160. № 2–3. P. 576. https://doi.org/10.1016/j.jhazmat.2008.03.028
Park K.-H., Balathanigaimani M.S., Shim W.-G., Lee J.-W., Moon H. // Microporous and Mesoporous Materials. 2010. V. 127. № 1–2. P. 1. https://doi.org/10.1016/j.micromeso.2009.06.032
Wang B., Zhu C., Zhang Z., Zhang W., Chen X., Sun N., Wei W., Sun Y., Ji H. // Fuel. 2016. V. 179. № 9. P. 274. https://doi.org/10.1016/j.fuel.2016.03.088
Kowalczyk P., Deditius A., Ela W.P., Wiśniewski M., Gauden P.A., Terzyk A.P., Furmaniak S., Włoch J., Kaneko K., Neimark A.V. // Carbon. 2018. V. 135. P. 12. https://doi.org/10.1016/j.carbon.2018.03.063
Lorenc-Grabowska E., Rutkowski P. // Applied Surface Science. 2014. V. 316. P. 435. https://doi.org/10.1016/j.apsusc.2014.08.024
Isaeva L.N., Tamarkina Y.V., Bovan D.V., Kucherenko V.A. // SibFU Journal. 2009. V. 2. № 1. P. 25. http://elib.sfu-kras.ru/handle/2311/1301
Fedorova N.I., Manina T.S., Ismagilov Z.R. // Solid Fuel Chemistry. 2015. V. 49. № 1. P. 30. https://doi.org/10.3103/S0361521915010048
Кучеренко В.А., Тамаркина Ю.В., Саберова В.А. // ХТТ. 2020. № 2. С. 22. [Solid Fuel Chemistry, 2020, vol. 54, no. 2, p. 79. https://doi.org/10.3103/S0361521920020068]https://doi.org/10.31857/S0023117720020061
Jagiello J., Olivier J.P. // Carbon. 2013. V. 55. P. 70. https://doi.org/10.1016/j.carbon.2012.12.011
Hadi P., Yeung K.Y., Barford J., An K.J., McKay G. // Chemical Engineering Journal. 2015. V. 269. P. 20. https://doi.org/10.1016/j.cej.2015.01.090
Li Y., Xing B., Wang X., Wang K., Zhu L., Wang S. // Energy Fuels. 2019. V. 33. № 12, P. 12459. https://doi.org/10.1021/acs.energyfuels.9b02924
Al-Ghouti M.A., Da’ana D.A. // Journal of Hazardous Materials. 2020. V. 393. P. 122383. https://doi.org/10.1016/j.jhazmat.2020.122383
Cazetta A.L., Vargas A.M.M., Nogami E.M., Kunita M.H., Guilherme M.R., Martins A.C., Silva T.L., Moraes J.C.G., Almeida V.C. // Chemical Engineering Journal. 2011. V. 174. № 1. P. 117. https://doi.org/10.1016/j.cej.2011.08.058
Mahadevi A.S., Sastry G.N. // Chem. Rev. 2013. V. 113. № 3. P. 2100. https://doi.org/10.1021/cr300222d
Дополнительные материалы отсутствуют.
Инструменты
Химия твердого топлива