

Журнал аналитической химии, 2022, T. 77, № 8, стр. 684-703
Применение спектроскопии гигантского комбинационного рассеяния для определения лекарственных и наркотических средств в биожидкостях человека
Н. Е. Маркина a, И. Ю. Горячева a, А. В. Маркин a, *
a Саратовский национальный исследовательский государственный университет
имени Н.Г. Чернышевского, Институт химии
410012 Саратов, Астраханская ул., 83, Россия
* E-mail: av_markin@mail.ru
Поступила в редакцию 24.06.2021
После доработки 30.07.2021
Принята к публикации 30.07.2021
- EDN: LESUGQ
- DOI: 10.31857/S0044450222080072
Аннотация
Обзор посвящен анализу возможностей, ограничений и особенностей применения спектроскопии гигантского комбинационного рассеяния света (ГКР) для определения лекарственных и наркотических средств в биологических жидкостях человека (БЖЧ) (кровь, моча, слюна). Анализ литературы показал, что в большинстве работ усилия исследователей направлены на преодоление основного недостатка ГКР как метода анализа – низкой селективности, что особенно актуально в случае таких сложных объектов, как биожидкости. В связи с этим основные положительные результаты в ГКР-анализе БЖЧ на настоящий момент связаны с разработкой методик, позволяющих минимизировать уровень фонового сигнала. Методики основаны на различных вариантах сочетания ГКР с методами разделения и концентрирования, использовании пробоподготовки, а также применении хемометрики для учета фоновой составляющей аналитического сигнала. Существенный прогресс также связан с созданием мультифункциональных ГКР-активных материалов, позволяющих улучшить селективность анализа. Однако именно аналитическая часть большинства работ является наиболее слабым местом, что требует дальнейших исследований, включая проверку методик ГКР-анализа контрольными методами.
Лекарственные (ЛС) и наркотические (НС) средства являются основными экзогенными компонентами биожидкостей человека (БЖЧ) (кровь, моча, слюна), подлежащими количественному определению. Необходимость определения ЛС в БЖЧ обусловлена развитием персонализированной медицины, одним из инструментов которой является терапевтический лекарственный мониторинг (ТЛМ) [1, 2]. Такой мониторинг необходим для достижения эффективного лечения пациентов и одновременного сведения к минимуму побочных эффектов, возникающих при применении ЛС с узким терапевтическим окном, например противоопухолевых и противоэпилептических препаратов. Также ТЛМ проводят для некоторых антибиотиков, поскольку их нерациональное использование вызывает токсическое действие на организм пациентов и образование новых резистентных штаммов бактерий. Определение НС в БЖЧ широко используется в судебно-медицинской экспертизе и при медицинском освидетельствовании с целью снижения количества криминальных случаев. Идентификация конкретного НС и его определение необходимы для проведения адекватного лечения и спасения жизни пациентов, пострадавших от передозировки неизвестным НС.
Определение ЛС и НС в БЖЧ является трудной задачей из-за сложного состава последних. Кроме того, концентрации основных компонентов БЖЧ (белки, мочевина, креатинин, мочевая кислота и др.) на порядок и более превышают содержание ЛС и НС, что приводит к низким соотношениям сигнал/шум. Помимо этого ЛС и НС могут связываться с белками и липидами БЖЧ, что дополнительно снижает концентрацию свободных молекул аналитов в пробах.
В настоящее время хромато-масс-спектрометрия является стандартным методом определения различных ЛС и НС в БЖЧ [3, 4]. Тем не менее высокая стоимость, трудоемкость, необходимость тщательной пробоподготовки и возможность работы только в стационарном варианте не позволяют широко использовать этот метод в экспресс-анализе и ТЛМ. Для обеспечения возможности экспресс-контроля в условиях лечебных учреждений активно разрабатывают портативные и экономичные системы. На сегодня такие системы чаще всего основаны на иммунохимических методах анализа и тест-методах на их основе [4, 5]. Главными преимуществами этих методов являются простота реализации, доступность портативных ридеров и высокая скорость анализа, что позволяет быстро определять концентрации аналитов на месте. В то же время необходимость разработки иммунореагентов для каждого конкретного аналита и зависимость селективности анализа от аффинности антител несколько ограничивают гибкость иммунохимических методов. Разработка и внедрение новых методов (био)химического экспрессного анализа все еще является актуальной задачей аналитической химии.
В качестве альтернативного метода определения ЛС и НС в БЖЧ все чаще предлагается спектроскопия гигантского комбинационного рассеяния света (ГКР) [6–8]. Спектроскопия ГКР основана на усилении интенсивности спектров комбинационного рассеяния (КР) молекул, находящихся вблизи металлических плазмонных наноструктур (ГКР-подложек), таких как металлические наночастицы (НЧ) и различные наноструктурированные материалы на их основе. Для изготовления ГКР-подложек чаще всего используют серебро и золото благодаря их высокой химической стабильности и возможности генерации поверхностного плазмонного резонанса в видимой области спектра [9].
Цель настоящего обзора − критический анализ публикаций, посвященных ГКР-определению ЛС и НС в БЖЧ, с целью оценки возможностей и ограничений спектроскопии ГКР. Для наиболее реального отражения уровня развития ГКР-методик для целей ТЛМ и наркоконтроля в обзоре проанализированы работы, посвященные ГКР-определению ЛС и НС в образцах реальных БЖЧ. Особое внимание в обзоре уделено анализу подходов, используемых для снижения влияния матричных эффектов БЖЧ.
ОСОБЕННОСТИ ПРИМЕНЕНИЯ СПЕКТРОСКОПИИ ГИГАНТСКОГО КОМБИНАЦИОННОГО РАССЕЯНИЯ ПРИ ОПРЕДЕЛЕНИИ ЛЕКАРСТВЕННЫХ И НАРКОТИЧЕСКИХ СРЕДСТВ В БИОЖИДКОСТЯХ ЧЕЛОВЕКА
Сравнение спектроскопии ГКР с другими методами анализа биожидкостей. Преимущества спектроскопии ГКР становятся очевидными при сравнении ее со стандартными методами определения ЛС и НС в БЖЧ. Аналогично хромато-масс-спектрометрии ГКР позволяет идентифицировать неизвестные аналиты, поскольку колебательные спектры содержат информацию о структуре определяемых молекул. Однако пробоподготовка образцов БЖЧ перед ГКР-анализом в основном проще. Также в настоящее время доступны портативные, относительно недорогие и простые в использовании КР-спектрометры, пригодные для проведения экспрессного анализа неспециалистами в условиях лечебных учреждений [6].
В отличие от иммунохимических методов, ГКР-анализ требует большего числа манипуляций, но без необходимости создания молекул-рецепторов для каждого аналита в отдельности. Более того, иммунореагенты имеют ограниченные сроки хранения, их создание требует времени и значительных финансовых затрат, что ограничивает возможности иммунохимических подходов в условиях быстрого внедрения большого числа новых структурно-родственных ЛС и НС. При проведении ГКР-анализа значительно легче устанавливать ложноположительные результаты, вызванные присутствием компонентов БЖЧ или нецелевых ЛС и НС благодаря различию профилей ГКР-спектров целевого аналита и мешающих соединений. Таким образом, по сравнению с иммунохимическим анализом спектроскопия ГКР является более универсальным инструментом, который можно адаптировать для определения различных веществ в разнообразных объектах с минимальными модификациями базовых реагентов (ГКР-подложек) и методик анализа.
Как и в случае других методов анализа, наибольшее негативное влияние на точность ГКР-определения ЛС и НС оказывает сложный состав БЖЧ. Так, присутствие внутренних компонентов БЖЧ может сильно влиять на ГКР-сигнал аналита, поскольку происходит усиление КР-сигнала любых молекул, находящихся вблизи ГКР-подложки [10]. Таким образом, затруднено ГКР-определение молекул аналита, которые слабее взаимодействуют с ГКР-подложкой по сравнению с компонентами БЖЧ и/или имеют слабый ГКР-сигнал.
Важно отметить, что в настоящее время активно ведутся работы по стандартизации метода ГКР, что в будущем позволит добиться его признания наравне с другими методами анализа [11, 12]. Например, проведена оценка межлабораторной воспроизводимости нескольких модельных методик ГКР-измерений в 15 разных лабораториях Европы, оснащенных КР-спектрометрами различных производителей (6 производителей) [11]. Результаты этого исследования показали, что относительное стандартное отклонение результатов измерений составило 12–13%, что является многообещающим, учитывая, что использовали различные спектрометры с различными параметрами настройки и сбора данных (мощность лазера, время накопления сигнала и т.д.).
Как и любой метод анализа, спектроскопия ГКР имеет ряд особенностей, которые могут как положительно, так и отрицательно влиять на результаты ГКР-анализа и должны учитываться и контролироваться при разработке новых методик определения аналитов в БЖЧ. Так, интенсивность ГКР-сигнала существенно зависит от экспериментальных условий (ионная сила, рН, длина волны и мощность возбуждающего излучения), а также от особенностей оптической системы прибора [13–15]. Для устранения проблем, связанных с воспроизводимостью ГКР-сигнала, предложено использование внутренних стандартов. Подход основан на нормировании интенсивности ГКР-сигнала аналита на интенсивность сигнала реагента, специально добавленного к анализируемой смеси и обладающего относительно интенсивным сигналом, при условии, что его спектр не перекрывается со спектром аналита [16–18].
Влияние величины рН на ГКР-сигнал. Следует отметить важность контроля уровня рН анализируемого раствора при измерении ГКР-спектров. Во-первых, рН среды определяет коллоидную и/или химическую стабильность ГКР-подложек на основе коллоидных растворов металлических наночастиц. Во-вторых, изменение величины рН значительно изменяет профиль и интенсивность ГКР-спектров молекул, поскольку (де)протонирование молекул приводит к изменению их поляризуемости (и, как следствие, интенсивности КР-сигнала), механизма адсорбции на поверхности ГКР-подложек и механизма усиления сигнала. Так, изменение рН может привести к значительному снижению интенсивности фонового ГКР-сигнала; например, креатинин и мочевая кислота имеют очень слабый ГКР-сигнал в кислой среде [19]. В-третьих, значение рН и буферная емкость БЖЧ могут варьироваться от образца к образцу, поэтому их следует учитывать при разработке методик анализа. Таким образом, контроль и корректировка величины рН позволяют управлять воспроизводимостью и интенсивностью ГКР-сигнала при разработке новой методики анализа.
Влияние длины волны возбуждающего излучения на интенсивность ГКР-сигнала. Другой особенностью ГКР-анализа является необходимость выбора оптимальной длины волны возбуждающего лазера. Этот параметр может влиять на результаты ГКР-измерений несколькими способами. Во-первых, длина волны лазера должна попадать в область длин волн, соответствующих поверхностному плазмонному резонансу материала ГКР-подложки. Например, для получения ГКР-спектров с использованием ГКР-подложек на основе меди длина волны возбуждающего лазера должна быть более 600 нм [20]. Во-вторых, при попадании длины волны лазера в диапазон электронных переходов аналита реализуется резонансное ГКР, которое имеет бóльшую интенсивность сигнала. Резонансное ГКР придает также дополнительную селективность по отношению к аналитам, для которых возможна реализация данного эффекта. Это можно наблюдать в случае некоторых ЛС, которые поглощают свет в видимой области спектра (например, доксорубицин [21], митоксантрон [22]). Однако некоторые компоненты БЖЧ (например, каротиноиды и метаболиты гемоглобина) также поглощают свет в видимой области спектра, тем самым увеличивая фоновый ГКР-сигнал за счет реализации резонансного ГКР. В-третьих, лазерное излучение может вызвать появление интенсивного сигнала флуоресценции, обусловленного наличием эндогенных флуорофоров БЖЧ. Для снижения интенсивности флуоресценции и ее влияния на ГКР-сигнал используют лазеры с максимально большой длиной волны излучения (>600 нм).
ВИДЫ ЛЕКАРСТВЕННЫХ И НАРКОТИЧЕСКИХ СРЕДСТВ
Анализ публикаций показал, что методики ГКР-определения ЛС и НС в БЖЧ разработаны для большого числа соединений, разнообразных по структуре (рис. 1). Наибольшее количество публикаций посвящено определению противоопухолевых ЛС [18, 22–34] (рис. 1а). Метод спектроскопии ГКР применяли также для определения представителей различных классов антибиотиков: цефалоспоринов [17, 35, 36], фторхинолонов [37, 38], пенициллинов [39], сульфаниламидов [19] и других природных [40] и синтетических [41] антибиотиков. Помимо противоопухолевых и антибактериальных ЛС, разработаны методики ГКР-определения представителей противоэпилептических [42–46], противопаркинсонических [47, 48], противогрибковых [49] и антигистаминных [50] ЛС, а также нейролептиков [16] и бета-адреномиметиков [51]. Представлены методики ГКР-определения следующих НС в БЖЧ: наиболее распространенных опиоидных наркотических анальгетиков [42, 44, 52–54], природных [43, 44, 55–60] и синтетических [43, 44, 61] НС, а также психостимуляторов [44, 62–67] (рис. 1б).
Рис. 1.
Основные виды лекарственных (а) и наркотических (б) средств, которые определяли в биожидкостях человека с использованием спектроскопии гигантского комбинационного рассеяния.

Важно отметить, что в некоторых публикациях разработаны методики определения нескольких представителей различных групп ЛС и НС разной химической природы [33, 42–44, 54]. Наибольшую универсальность, т.е. возможность одновременного определения более четырех разнородных НС и ЛС, продемонстрировали методики, основанные на сочетании ГКР с ВЭЖХ [42] и твердофазной экстракцией [43, 44].
Актуальным направлением является разработка методик ГКР-анализа с учетом структурной родственности аналитов. Так, имеются работы по определению в БЖЧ нескольких представителей цефалоспориновых антибиотиков [17, 36], бензодиазепинов [45, 46], психостимуляторов [66] и природных НС [55]. Однако в большинстве опубликованных исследований представлены методики, которые разработаны и протестированы с использованием только одного аналита (табл. 1). Трудности разработки универсальных методик определения даже представителей ЛС и НС одного класса связаны с тем, что изменение функциональных групп молекул аналита может значительно изменить их ориентацию и степень адсорбции на поверхности ГКР-подложки. Такие изменения, в свою очередь, часто влияют на интенсивность и профиль ГКР-спектра, приводя к необходимости корректировки методики пробоподготовки для каждого конкретного аналита. Дополнительно отметим, что пока нет работ по одновременному ГКР-определению нескольких ЛС или НС в БЖЧ. Таким образом, актуальным направлением является повышение универсальности и мультиплексности методик ГКР-анализа с учетом природы и класса ЛС или НС, а также вида БЖЧ.
Таблица 1.
Примеры методик ГКР-определения лекарственных и наркотических средств в биожидкостях человека, сгруппированные по подходам и методам, использованным для улучшения точности анализа
| Подходы и методы | БЖЧ (количество образцов) |
Аналит | ГКР-подложка (λex, нм) | ПрО, мкМ (наличие ГГ) | Лите-ратура |
|---|---|---|---|---|---|
| ГКР-определение ЛС и НС в БЖЧ без использования методов разделения и концентрирования | |||||
| Математическая обработка результатов анализа (применение хемометрики) | Разбавленная плазма крови (1) | Паклитаксел | Полистирольные микрочастицы с золотым покрытием (785) | 0.004 (+) | [25] |
| Разбавленная сыворотка крови (н.д.) | Метотрексат | Фильтровальная бумага, покрытая НЧ золота (785) | – (+) | [27] | |
| Плазма крови (6*) | Иматиниб | Кремниевая подложка с серебряным покрытием (785**) | 0.43 (+) | [30] | |
| Моча (3) | Моксифлоксацин | Кремниевая подложка с золотым покрытием (785) | 65 (+) | [37] | |
| Разбавленная моча (9) | Левофлоксацин | Гидроксиламиновые НЧ серебра в микрофлюидной системе (532**) | – (+) | [38] | |
| Плазма крови (1) | Кодеин | Цитратные НЧ серебра (633**) | 1.39 (+) | [52] | |
| Слюна (1) | Кокаин | Покрытие из наноразмерных дендритов серебра (633) | – (–) | [57] | |
| Моча (50*) | Метамфетамин | Золотые наностержни, стабилизированные ЦТАБ (785**) | – (–) | [63] | |
| Модификация поверхности ГКР-подложек | Плазма крови (н.д.) | Доксорубицин | Покрытие из золота, модифицированное самособирающимся монослоем поликарбоксибетаина (785) | – (+) | [28] |
| Плазма крови (10) | Эрлотиниб | НЧ золота, покрытые модификатором с азидной группой (785) | – (+) | [32] | |
| Сыворотка крови (н.д.) | Паклитаксел, циклофосфамид | Покрытие из золотых нанозвезд, модифицированных самособирающимся монослоем L-цистеина (785) | – (+) | [33] | |
| Простая пробоподготовка | Моча (7) | Цефтриаксон | Цитратные НЧ меди (638**) | 13 (+) | [17] |
| Цефазолин | 19 (+) | ||||
| Цефоперазон | 56 (+) | ||||
| Сыворотка крови (н.д.) | Митоксантрон | Цитратные НЧ серебра в проточной системе (633) | 4 × 10–5 (+) | [22] | |
| Слюна (н.д.) | 5-Фторурацил | Покрытие из НЧ серебра внутри стеклянного капилляра (785) | 1.15 (+) | [24] | |
| Цельная кровь (1) | 6-Меркаптопурин | Игла, модифицированная наноструктурами ядро-оболочка на основе золота (785) | – (–) | [26] | |
| Простая пробоподготовка | Разбавленная сыворотка крови (н.д.) | 6-Тиогуанин | Цитратные НЧ золота в микрофлюидной системе (638) | 0.66 (+) | [34] |
| Моча (6*) | Берберин | Цитратные НЧ серебра (633) | 0.0016 (+) | [40] | |
| Разбавленная моча (10) | Нитроксолин | Гидроксиламиновые НЧ серебра в микрофлюидной системе (532) | 3.43 (+) | [41] | |
| Плазма крови (15*) | Допамин | Покрытие из НЧ серебра на оптически прозрачной токопроводящей подложке (633**) | – (+) | [48] | |
| Моча (7*) | Цетиризин | Цитратные НЧ серебра (785) | 0.013 (+) | [50] | |
| Разбавленная слюна (1) | Метамфетамин | Цитратные НЧ серебра в микрофлюидной системе (633) | – (–) | [62] | |
| Моча (1) | Метамфетамин, амфетамин | Кремниевая подложка с серебряным покрытием (785**) | – (–) | [66] | |
| ГКР-определение ЛС и НС в БЖЧ с привлечением методов разделения и концентрирования | |||||
| ВЭЖХ | Моча (4*) | Метотрексат | Цитратные НЧ серебра (785**) | 2.36 (+) | [29] |
| 7-гидроксиметотрексат | 1.84 (+) | ||||
| 2,4-диамино-N(10)-метилптериновая кислота | 3.26 (+) | ||||
| Моча (2*), кровь (1*) | Дигидрокодеин, кодеин, доксепин, циталопрам, карбамазепин, метадон | Пластиковая подложка с серебряным покрытием (514) | – (–) | [42] | |
| ТСХ | Моча (1) | Клозапин | Цитратные НЧ серебра (785**) | 0.3 (+) | [16] |
| N-десметилклозапин | – (–) | ||||
| Клозапин N-оксид | – (–) | ||||
| Плазма крови (1) | Апоморфин | Цитратные НЧ серебра (785) | – (–) | [47] | |
| Моча (3*) | Котинин | Цитратные НЧ серебра (647) | 0.01 (+) | [55] | |
| Гидроксикотинин | 0.2 (+) | ||||
| Плазма крови (н.д.) | Кокаин | Цитратные НЧ золота (785) | 0.033 (+) | [58] | |
| Моча (1*) | Тетрагидроканнабинол | Цитратные НЧ золота (785**) | – (–) | [59] | |
| Электрофорез | Слюна (2*) | Параксантин | Серебряный электрод с покрытием из НЧ серебра (1064) | – (–) | [67] |
| ЖЖЭ | Моча (13) | Сульфаметоксазол | Гидроксиламиновые НЧ серебра (488) | 6.7 (+) | [19] |
| Моча (1) | Флунитразепам, диазепам, нордиазепам | Цитратные НЧ золота (785) | 0.006–0.037 (+) | [45] | |
| Моча (н.д.) | 7-аминофлунитразепам | Цитратные НЧ золота (785) | 0.031 (+) | [46] | |
| Алпразолам | 3.56 (+) | ||||
| Хлордиазепоксид | 4.68 (+) | ||||
| Клоназепам | 2.82 (+) | ||||
| Диазепам | 0.019 (+) | ||||
| Флунитразепам | 0.128 (+) | ||||
| Лоразепам | 4.67 (+) | ||||
| Мидазолам | 33.8 (+) | ||||
| Нордиазепам | 1.551 (+) | ||||
| Оксазепан | 0.837 (+) | ||||
| Триазолам | 20.4 (+) | ||||
| Моча (15*) | Кленбутерол | Композит НЧ золота с оксидом графена (785**) | 0.0018 (+) | [51] | |
| Разбавленная моча (5*) | Морфин | Золотые наностержни, стабилизированные ЦТАБ (785**) | – (–) | [53] | |
| Разбавленная моча (1) | Кокаин | НЧ золота, стабилизированные ЦТАБ (785**) | – (–) | [56] | |
| Слюна (н.д.) | Кокаин | Планарные коммерческие ГКР-подложки (785) | – (–) | [60] | |
| Моча (30) | Метамфетамин | Золотые наностержни, стабилизированные ЦТАБ (785**) | – (–) | [64] | |
| Моча (6) | Метамфетамин | Эмульсия масло-в-воде с НЧ серебра (532) | – (–) | [65] | |
| ТФЭ | Моча (11) | Метотрексат | Порошок оксида алюминия со встроенными НЧ серебра (532**) | 9.3 (+) | [18] |
| Слюна (н.д.) | 5-Фторурацил | Силикатный золь-гель материал со встроенными НЧ серебра (785) | 15.4 (–) | [23] | |
| Сыворотка крови (1) | 6-Тиогуанин | Альгинатные микрочастицы со встроенными НЧ золота (638) | – (–) | [31] | |
| Плазма крови (н.д.) | Клоксациллин | Кремниевая подложка с золотым покрытием (780) | – (–) | [39] | |
| Слюна (н.д.) | Кокаин, фенциклидин, диазепам, парацетамол | Силикатный золь-гель материал со встроенными НЧ золота (785) | – (–) | [43] | |
| Слюна (н.д.) | Кокаин, амфетамин, диазепам, метадон, фенциклидин | Силикатный золь-гель материал со встроенными НЧ золота (785) | – (–) | [44] | |
| Разбавленная слюна (1) | Синтетический каннабиноид | Цитратные НЧ золота (785) | 0.077 (+) | [61] | |
| НТФЭ | Моча (5) | Цефтриаксон | Гидроксиламиновые НЧ серебра (473) | 0.7 (+) | [35] |
| Моча (5) | Цефазолин | Гидроксиламиновые НЧ серебра (638**) | 12.8 (+) | [36] | |
| Цефоперазон | 21.1 (+) | ||||
| Цефотаксим | 22.6 (+) | ||||
| Цефтриаксон | 50.3 (+) | ||||
| Цефуроксим | 199.8 (+) | ||||
| Сыворотка крови (1) | Флуцитозин | Нитроцеллюлозные мембраны с нанесенными НЧ серебра (785#) | – (+) | [49] | |
| Слюна (1), плазма крови (1), кровь (1) | Кодеин, фентанил | Тест-полоска с ГКР-активной областью из НЧ золота (785**) | – (–) | [54] | |
Обозначения: БЖЧ – биологическая жидкость человека; ПрО – предел обнаружения; λex – длина волны лазерного излучения, использованного для генерации ГКР-сигнала; ГГ – градуировочный график; ЛС – лекарственные средства; НС – наркотические средства; н.д. – нет данных; ТСХ – тонкослойная хроматоргафия; ЖЖЭ – жидкостно-жидкостная экстракция; ТФЭ – твердофазная экстракция; НТФЭ – неудерживающая твердофазная экстракция; ЦТАБ – цетилтриметиламмония бромид.
ВИДЫ БИОЖИДКОСТЕЙ
Наибольшее количество методик ГКР-определения ЛС и НС разработано для образцов мочи (примерно половина работ, рис. 2). Такая популярность данной БЖЧ обусловлена простотой и неинвазивностью ее сбора. Однако важно отметить, что разработка методик определения ЛС именно в крови (плазме или сыворотке) является наиболее актуальной задачей ТЛМ (рис. 2а), поскольку терапевтический эффект конкретного ЛС напрямую зависит от его концентрации именно в крови пациента. Для определения НС популярными БЖЧ являются моча и слюна, пригодные для неинвазивного и оперативного скрининга потребления НС (рис. 2б).
Рис. 2.
Распределение разработанных методик ГКР-определения лекарственных и наркотических средств по типам объектов анализа.

Несмотря на достаточно большое число публикаций, посвященных работе с образцами реальных БЖЧ (табл. 1), лишь в двух работах [42, 54] предложены достаточно универсальные методики ГКР-анализа, пригодные для определения НС в различных БЖЧ. Так, авторы показали возможность регистрации ГКР-спектров нескольких НС в моче и в крови [42], а также в моче и в слюне [54]. Значительная часть работ, приведенных в табл. 1, имеет существенные методологические недостатки, снижающие ценность полученных результатов. Так, примерно в 20% работ образцы БЖЧ разбавляли чистой водой или буферным раствором перед введением добавки аналита. Использование такого приема свидетельствует о недостаточной селективности предложенных методик при высокой концентрации внутренних компонентов БЖЧ.
Более чем в половине работ анализировали только один образец БЖЧ, либо данные о количестве использованных образцов отсутствовали (табл. 1). Это ставит под сомнение полученные результаты, поскольку в этих случаях не учитывалась вариативность состава БЖЧ, например, от человека к человеку или в зависимости от времени сбора образцов.
Тем не менее примеры корректных методик ГКР-определения ЛС и НС (в том числе с учетом вариативности состава БЖЧ) [17–19, 29, 30, 35–37, 40, 50, 51, 55] показывают принципиальную возможность нивелирования отрицательного влияния сложного состава БЖЧ на точность ГКР-анализа. Для достижения максимального контроля над ГКР-сигналом при анализе БЖЧ требуется дополнительное изучение взаимодействий металлической ГКР-активной поверхности или аналита с компонентами БЖЧ и их влияния на ГКР-сигнал.
ПОДЛОЖКИ, ПРИМЕНЯЕМЫЕ В СПЕКТРОСКОПИИ ГИГАНТСКОГО КОМБИНАЦИОННОГО РАССЕЯНИЯ
Эффект ГКР реализуется только при адсорбции молекул аналита на поверхности ГКР-подложки. Рассмотрим виды подложек, применяемых для определения ЛС и НС в БЖЧ, их преимущества и ограничения. Вопросам создания и изучения ГКР-подложек посвящено большое число оригинальных и обзорных статей [9, 68], поэтому в данном разделе рассмотрим лишь самые общие моменты, а некоторые важные и интересные примеры будут рассмотрены ниже.
ГКР-подложки, применяемые для регистрации спектров ЛС и НС в БЖЧ, можно условно разделить на три группы: коллоидные НЧ, планарные подложки и мультифункциональные композиты (табл. 1). Примерно в половине работ для ГКР-анализа использовали коллоидные НЧ металлов, и из них наиболее популярными оказались НЧ, стабилизированные цитрат-ионами, что обусловлено простотой и воспроизводимостью их синтеза. В качестве других стабилизаторов использовали гидроксиламин и ЦТАБ, причем важным преимуществом последнего стабилизатора является возможность его использования при ГКР-определении гидрофобных соединений, например морфина [53].
Планарные ГКР-подложки использовали примерно в трети работ, и их главным элементом является наноструктурированная ГКР-активная поверхность. Такие подложки получают либо огрублением металлической поверхности до получения наноразмерной шероховатости, либо нанесением ГКР-активного слоя на какую-либо поддерживающую матрицу. Поддерживающую матрицу также иногда используют для задания и/или дополнительного контроля морфологии поверхности конечной ГКР-подложки. Наиболее распространенными вариантами получения планарных ГКР-подложек являются нанесение заранее синтезированных коллоидных НЧ на твердую основу [27, 33, 49, 54], электрохимическая обработка токопроводящей поверхности [48, 57] и физическое нанесение ГКР-активного слоя из вакуума на подложки из кремния или других материалов [25, 37, 39, 60]. Важно, что в случае электрохимического и вакуумного нанесения получают ГКР-активный слой без использования дополнительных стабилизирующих агентов, которые могут формировать фоновый ГКР-сигнал.
Мультифункциональные композитные ГКР-подложки используют реже, их главным отличием от двух предыдущих типов подложек является наличие дополнительного компонента, служащего для значительного улучшения результатов анализа. Например, композитными ГКР-подложками являются ГКР-активные сорбенты, состоящие из (не)органических сорбентов со встроенными металлическими НЧ. Матрица сорбента придает подложке большую сорбционную емкость, а в некоторых случаях дополнительно отделяет молекулы аналита от компонентов БЖЧ.
В качестве материала ГКР-активных участков всех типов подложек в равной степени используют золото и серебро. Наночастицы меди при ГКР-определении ЛС в БЖЧ использовали только в одной работе [17]. Важно отметить, что в данной работе НЧ меди продемонстрировали эффективность усиления КР-спектров некоторых цефалоспориновых антибиотиков, сравнимую с таковой для НЧ на основе серебра и золота. Также, несмотря на низкую стабильность медных НЧ во времени (окисление и потеря усиливающих свойств происходит в течение получаса), авторы показали, что использование свежеприготовленных НЧ меди позволяет достичь воспроизводимого ГКР-сигнала.
Однако, несмотря на высокий уровень развития подходов, связанных с получением и изучением ГКР-подложек, создание “универсальной” подложки для определения ЛС и НС в БЖЧ невозможно. Это связано со значительным различием степени взаимодействия разных аналитов с ГКР-подложками различного состава, что напрямую сказывается на интенсивности и воспроизводимости аналитического сигнала и на вероятности возникновения конкурентных взаимодействий с мешающими компонентами. В связи с этим перспективным направлением является разработка базовых ГКР-подложек, ориентированных на отдельные типы БЖЧ (с учетом присутствия основных мешающих веществ) и группы аналитов (с учетом особенностей их строения). В этом случае главной задачей является достижение компромиссной универсальности, способствующей определению аналитов внутри группы при минимальной корректировке базовой методики.
Другой проблемой, связанной с ГКР-подложками, является отсутствие стандартизации при оценке их аналитических характеристик. Так, на настоящий момент отсутствуют готовые методики, позволяющие объективно сравнивать аналитическую эффективность различных ГКР-подложек. Об отсутствии единого мнения относительно того, какие подложки работают лучше, также свидетельствует большое разнообразие использованных подложек (табл. 1). Таким образом, разработка и апробация методик анализа с использованием доступных, стандартизированных и недорогих ГКР-подложек и сегодня является актуальной задачей для обеспечения возможности применения ГКР вне исследовательских лабораторий.
ПРИМЕНЕНИЕ СПЕКТРОСКОПИИ ГИГАНТСКОГО КОМБИНАЦИОННОГО РАССЕЯНИЯ ДЛЯ ОПРЕДЕЛЕНИЯ ЛЕКАРСТВЕННЫХ И НАРКОТИЧЕСКИХ СРЕДСТВ В БИОЖИДКОСТЯХ ЧЕЛОВЕКА БЕЗ ИСПОЛЬЗОВАНИЯ МЕТОДОВ РАЗДЕЛЕНИЯ И КОНЦЕНТРИРОВАНИЯ
Для достижения максимальной эффективности ГКР при определении ЛС и НС в БЖЧ предложено использование различных подходов, в основном нацеленных на увеличение селективности анализа путем уменьшения влияния мешающих компонентов (эндо- и экзогенных компонентов БЖЧ) на аналитический сигнал. В данном обзоре все методики ГКР-анализа условно разделены на две группы: без и с разделением молекул аналита и мешающих компонентов перед ГКР-определением.
Ниже рассмотрим методики без предварительного отделения аналитов, основанные на использовании: (i) обработки аналитического сигнала с помощью многомерного анализа данных (хемометрики) для снижения влияния фонового сигнала на результаты анализа; (ii) простых способов пробоподготовки (например, изменение рН и разбавление), которые изменяют силу взаимодействия ГКР-подложек с молекулами аналита и/или мешающими компонентами; (iii) модификации поверхности ГКР-подложек молекулами-модификаторами, изменяющими взаимодействие между подложкой и аналитом, а также примесями.
Обработка результатов ГКР-измерений методами хемометрики. Использование подходов хемометрики для учета фонового сигнала и нивелирования его влияния на точность результатов ГКР-анализа БЖЧ − весьма популярное направление [25, 27, 30, 37, 38, 52, 57, 63]. Повышение точности и селективности анализа в таком случае достигается за счет, во-первых, предварительной обработки зарегистрированных ГКР-спектров и удаления вкладов рэлеевского рассеяния и флуоресценции, а также спектральных артефактов и промахов и, во-вторых, использования в качестве аналитического сигнала всего профиля ГКР-спектра аналита вместо какого-либо единичного пика. Одним из преимуществ применения хемометрики является возможность упрощения или даже исключения тщательной пробоподготовки образцов, ограничиваясь, например, только разбавлением образцов и/или удалением белков [30, 52, 57]. Эффективность использования хемометрики подтверждена в некоторых работах путем валидации построенной математической модели контрольными методами с использованием клинических образцов [30, 63]. Однако данный подход имеет ограниченную применимость (вплоть до его полной непригодности) в случаях слабого собственного ГКР-сигнала аналита, или когда мешающие компоненты БЖЧ подавляют взаимодействие аналита с ГКР-подложкой, что может привести к полному исчезновению аналитического сигнала и необходимости использования дополнительной пробоподготовки.
Модификация поверхности ГКР-подложек. Модифицирование ГКР-активных поверхностей молекулами, улучшающими селективность ГКР-анализа, предложено для отделения целевых аналитов от компонентов БЖЧ. Например, для предотвращения блокировки ГКР-активной поверхности белками плазмы использовали модифицирование поверхности ГКР-подложки самособирающимся монослоем цвиттер-ионного ПАВ [28]. Разработанную подложку интегрировали в проточную систему, что позволило определять концентрацию доксорубицина в плазме крови без пробоподготовки и максимально автоматизированно. Аналогичный подход использовали для определения противоопухолевых ЛС в сыворотке крови [33]. При этом поверхность ГКР-подложки покрывали самособирающимся монослоем аминокислоты (L-цистеином), что позволило уменьшить степень сорбции белка на модифицированной подложке примерно на 80%.
Другим направлением является модификация поверхности ГКР-подложки молекулами, которые вступают с аналитом в химическую реакцию, приводящую либо к образованию продукта с интенсивным ГКР-сигналом [32, 69], либо к снижению ГКР-сигнала исходной молекулы-модификатора (КР-маркера) [70]. Например, разработана методика конкурентного формата определения противоопухолевого препарата эрлотиниба в плазме крови [32], основанная на химической реакции между азидной группой модификатора ГКР-подложки и алкинной группой аналита или конкурирующего КР-маркера. Несмотря на трудоемкость и длительность (время анализа достигает одного часа), предложенный авторами подход позволяет проводить ГКР-определение веществ, не обладающих собственным интенсивным ГКР-сигналом.
Несмотря на перспективность применения разнообразных селективных модификаторов (аптамеры, циклодекстрины, пептиды, полимеры с молекулярными отпечатками и др.), возможность их использования в ГКР-определении ЛС и НС в реальных БЖЧ пока не изучена. Тем не менее предложена методика ГКР-определения биомаркера опухолевого роста (ацетил амантадин) в искусственной моче с применением ГКР-подложки, поверхность которой модифицирована молекулами β-циклодекстринов [71]. Супрамолекулярные взаимодействия позволяют также контролировать ориентацию молекул аналитов по отношению к поверхности ГКР-подложки [72]. Данная особенность имеет важное значение, поскольку известно, что помимо прочего ГКР-сигнал зависит от ориентации молекул относительно электромагнитных полей, возникающих при генерации поверхностного плазмонного резонанса.
Применение простой пробоподготовки. Большинство работ по ГКР-определению ЛС и НС в БЖЧ основано на простом смешивании образца БЖЧ с ГКР-подложкой [17, 22, 24, 26, 34, 40, 41, 48, 50, 62, 66]. Однако при этом молекулы аналитов должны обладать большей аффинностью к поверхности ГКР-подложки или более интенсивным ГКР-сигналом по сравнению с молекулами компонентов БЖЧ. Для дополнительного повышения точности результатов в таких методиках часто используют простые этапы пробоподготовки БЖЧ, такие как разбавление образца [17, 40], корректировку его рН [66], а также (ультра)фильтрацию для удаления белков и клеточных компонентов БЖЧ (особенно в случаях плазмы и сыворотки крови) [34, 40, 41, 50, 62].
Анализ литературы показал, что в качестве ГКР-подложек в большинстве работ с простой пробоподготовкой используют коллоидные НЧ (табл. 1). Кроме того, в качестве ГКР-подложек предложены инъекционные иглы [26] и стеклянные капилляры [24] для забора крови с нанесенным ГКР-активным слоем, что позволяет уменьшить объем используемой БЖЧ и совместить процедуру отбора проб БЖЧ и ГКР-определение аналита. Например, продемонстрирована возможность in vivo определения 6-меркаптопурина в цельной крови путем введения ГКР-активной иглы в ушную вену кролика [26].
Важно отметить, что при использовании коллоидных НЧ в качестве ГКР-подложек часто необходима их искусственная агрегация для дополнительного усиления ГКР-сигнала [73]. Однако процесс агрегации является динамическим, что может снижать воспроизводимость результатов ГКР-анализа. Более того, некоторые аналиты и компоненты БЖЧ (в первую очередь, неорганические соли) также могут способствовать агрегации НЧ. Особенно ярко эта проблема может проявляться при анализе мочи из-за ее переменчивого состава. Популярным подходом к улучшению контроля над агрегацией и повышению воспроизводимости ГКР-сигнала является использование проточных (в том числе микрофлюидных) систем. Данные системы обеспечивают хорошо контролируемую и автоматизированную агрегацию коллоидных НЧ и/или корректировку уровня рН образцов [22, 34, 41, 62]. Тем не менее использование микрофлюидных систем не позволяет уменьшить фоновый ГКР-сигнал или отделить молекулы аналита от компонентов БЖЧ, а также требует применения довольно сложного и дорогостоящего оборудования.
Важным, но недооцененным этапом разработки методик ГКР-определения ЛС и НС в БЖЧ, основанных на простом смешивании образца БЖЧ и ГКР-подложки, является исследование влияния отдельных компонентов БЖЧ и их смесей на ГКР-сигнал аналита. Указанный этап позволяет выявить конкретные вещества, приводящие к увеличению фонового ГКР-сигнала или уменьшению сигнала целевого аналита. Например, в работе, посвященной определению цефалоспориновых антибиотиков в моче [17], показано, что даже патологически высокие концентрации некоторых внутренних компонентов мочи могут лишь незначительно влиять на интенсивность ГКР-сигнала аналитов. Это продемонстрировано с использованием пятикратного искусственного увеличения содержания мочевины, креатинина, глюкозы или белков относительно нормальных значений. Однако увеличение ионной силы мочи за счет добавления смеси солей, обычно входящих в ее состав, привело к снижению интенсивности ГКР-сигнала антибиотиков. Также установлено, что одновременное превышение концентрации перечисленных органических и неорганических компонентов в моче влияет на ГКР-сигнал так же, как и увеличение только ионной силы. Таким образом, авторы показали, что неорганические компоненты БЖЧ и создаваемая ими ионная сила могут значительно сильнее влиять на точность ГКР-анализа по сравнению с более КР-активными компонентами БЖЧ, такими как органические молекулы. В другой работе, посвященной определению 6-тиогуанина в сыворотке крови, авторы предварительно проверили влияние некоторых компонентов сыворотки (белок, основные анионы и катионы, а также некоторые серосодержащие аминокислоты) на ГКР-сигнал аналита [34]. Результаты показали, что для уменьшения негативного влияния аминокислот на ГКР-сигнал 6-тиогуанина достаточно откорректировать рН до значения 8, а для устранения влияния белков провести ультрафильтрацию образцов сыворотки крови. Таким образом, селективность разработанных ГКР-методик по отношению к определенному аналиту по сравнению с основными компонентами БЖЧ изучена и подтверждена лишь в нескольких работах.
ПРИМЕНЕНИЕ СПЕКТРОСКОПИИ ГИГАНТСКОГО КОМБИНАЦИОННОГО РАССЕЯНИЯ ДЛЯ ОПРЕДЕЛЕНИЯ ЛЕКАРСТВЕННЫХ И НАРКОТИЧЕСКИХ СРЕДСТВ В БИОЖИДКОСТЯХ ЧЕЛОВЕКА С ПРИВЛЕЧЕНИЕМ МЕТОДОВ РАЗДЕЛЕНИЯ И КОНЦЕНТРИРОВАНИЯ
Высокоэффективная жидкостная хроматография (ВЭЖХ). Спектроскопия ГКР предложена в качестве метода детектирования при разделении методом ВЭЖХ. Однако такое сочетание методов требует больших затрат труда и времени, поскольку замена УФ- или масс-селективного детектирования на ГКР не исключает необходимости тщательной пробоподготовки образцов БЖЧ перед разделением. Особое внимание при этом следует уделять выбору подвижной фазы, поскольку некоторые растворители (например, ацетонитрил) могут конкурировать с аналитом за сорбционные центры на поверхности ГКР-подложки, что приводит к подавлению ГКР-сигнала аналита или возникновению интенсивного фонового сигнала. Помимо этого, состав подвижной фазы может влиять на растворимость аналита, что приводит к уменьшению его сродства к ГКР-подложке. Наконец, подвижная фаза может влиять на коллоидную стабильность некоторых ГКР-подложек, снижая воспроизводимость результатов анализа.
Несмотря на перечисленные ограничения, немногочисленные работы по разработке методик ВЭЖХ-ГКР-определения ЛС и НС в БЖЧ [29, 42] четко демонстрируют возможность эффективной идентификации неизвестных аналитов благодаря разделению компонентов образца и многополосной структуре ГКР-спектров. Так, с помощью ВЭЖХ-ГКР идентифицированы НС в крови и моче наркозависимых пациентов [42] и показано, что в некоторых фракциях, соответствующих появлению пиков на хроматограмме (согласно результатам ВЭЖХ-УФ), содержатся смеси НС, их метаболитов и продуктов разложения, причем состав этих фракций можно различить по ГКР-спектрам. Также установлено, что метаболиты и/или продукты распада структурно-родственных НС имеют в ГКР-спектрах некоторые одинаковые пики, что важно при разработке универсальных методик анализа.
В работе [29] по применению сочетания ГКР и ВЭЖХ показана возможность определения не только основного ЛС (метотрексат) в моче, но и его метаболитов. Разработанная ВЭЖХ-ГКР-методика валидирована с использованием контрольного метода (ВЭЖХ-УФ) и образцов мочи пациентов, принимавших метотрексат для лечения артрита.
Стоит отметить, что авторами указанных работ обсуждена полезность ВЭЖХ-ГКР как метода получения дополнительной информации о неизвестных аналитах/метаболитах, которую затруднительно или нельзя получить при использовании других детекторов. Таким образом, авторы предлагают использовать ВЭЖХ-ГКР как дополнительный метод анализа при проведении, например, фармакокинетических исследований и судмедэкспертизы.
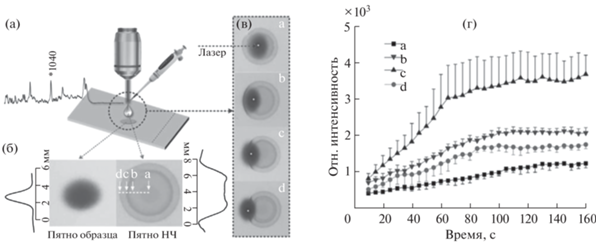
Тонкослойная хроматография (ТСХ). Методики, основанные на сочетании ТСХ и ГКР, включают разделение смеси на ТСХ-пластине, добавление НЧ металла к зоне аналита и детектирование ГКР-сигнала в данной зоне [16, 47, 55, 58, 59]. В качестве стационарной фазы используют ТСХ-пластины с силикагелем [16, 47, 55] или диатомитом [58, 59]. Хорошим примером использования ТСХ-пластин с силикагелем является работа по ТСХ-ГКР-определению клозапина в моче [16]. Поскольку после добавления НЧ серебра к зоне ЛС на пластине ТСХ происходит размытие пятна аналита, а также радиальная диффузия и неравномерное распределение НЧ на поверхности пластины (рис. 3), авторы работы провели поиск оптимального участка для ГКР-детектирования и получения максимально воспроизводимого и интенсивного сигнала. Для увеличения воспроизводимости результатов анализа авторы добавляли к образцам мочи внутренний стандарт (раствор NaSCN), ГКР-сигнал которого использовали для нормировки аналитического сигнала. Результаты тестирования предложенной методики показали, что ГКР-определение аналита на пластине ТСХ примерно на порядок чувствительнее по сравнению с УФ-визуализацией, а также позволяет детектировать метаболиты клозапина.
Рис. 3.
(а): Схематическое изображение методики определения, основанной на сочетании тонкослойной хроматографии с ГКР-детектированием (ТСХ-ГКР). (б): Денситограмма пятна образца и пятна наночастиц серебра под УФ-излучением (254 нм); стрелками а–d показаны различные позиции регистрации ГКР-сигнала. (в): Изображения пластины ТСХ с указанием места регистрации ГКР-сигнала (белая точка). (г): Зависимость интенсивности ГКР-сигнала на пластине ТСХ от места регистрации (a–d) и времени взаимодействия наночастиц серебра с молекулами аналита. Адаптировано из статьи [16] с разрешения правообладателя.
В работе [55] по ТСХ-ГКР-определению метаболитов никотина в моче пришлось дополнительно использовать жидкостно-жидкостную экстракцию (ЖЖЭ) аналитов из мочи перед ТСХ-разделением, чтобы достичь нужной чувствительности анализа. После разделения авторы соскабливали силикагель с пластины ТСХ в зоне аналита и элюировали молекулы аналитов перед ГКР-определением. Несмотря на значительное увеличение продолжительности анализа из-за включения дополнительных этапов, достигнутые пределы обнаружения позволили определять метаболиты никотина в моче не только активных, но и пассивных курильщиков.
Перспективно использование пластин ТСХ, модифицированных полимерами с молекулярными отпечатками, для увеличения селективности разделения и определения аналитов [74]. Другое интересное направление − применение для пластины ТСХ неорганического сорбента, предварительно модифицированного ГКР-активными элементами [75, 76]. Такой подход усложняет приготовление ГКР-активной ТСХ-пластины, но минимизирует количество манипуляций непосредственно при проведении анализа. В данном случае не требуется поиск места регистрации аналитического ГКР-сигнала на пластине ТСХ, поскольку НЧ равномерно распределены на ее поверхности. Следует подчеркнуть, что отмеченные направления пока не реализованы для определения ЛС и НС в БЖЧ.
Во всех перечисленных выше работах для регистрации ГКР-сигнала с пластины ТСХ использовали лазеры с эмиссией в красном и ближнем ИК-диапазоне (647 и 785 нм) (табл. 1), что обусловлено следующими причинами. Во-первых, при использовании лазеров с большей энергией фотонов (473–532 нм) увеличивается вероятность “горения” молекул аналита из-за быстрого высыхания матрицы сорбента. В результате в ГКР-спектре исчезают пики аналита и появляются интенсивные и широкие пики при 1350 и 1600 см–1, соответствующие элементарному углероду, что дополнительно усложняет анализ. Во-вторых, фотоны синей и зеленой областей спектра могут приводить к появлению сильного фонового сигнала в результате возбуждения сигнала флуоресценции индикатора пластины ТСХ из-за высокой плотности мощности лазерного излучения.
Электрофоретическое разделение. Интеграция электрофоретического (ЭФ) разделения и ГКР заключается в использовании электрохимической ячейки для разделения заряженных эндо- и экзогенных компонентов БЖЧ и их принудительной электростатической сорбции на ГКР-активном электроде, используемом далее в качестве ГКР-подложки для регистрации сигнала. Единственным примером использования ЭФ-ГКР для анализа реальной БЖЧ является работа [67] по определению в слюне параксантина (основного метаболита кофеина). Авторы определили оптимальное значение поляризации ГКР-активного электрода, при котором достигается максимальный ГКР-сигнал аналита, однако варьирование величины поляризации не позволило устранить фоновый ГКР-сигнал слюны, обусловленный присутствием фенилаланина. Тем не менее пик ГКР параксантина, по интенсивности которого проводили определение, не перекрывается с ГКР-пиками фонового сигнала слюны, что позволило установить корреляцию между дозой и временем после приема кофеина и его концентрацией в слюне.
Опубликованы работы по ЭФ-ГКР-определению некоторых ЛС и НС в чистой воде [77] и в искусственных БЖЧ [78, 79], результаты которых можно попытаться экстраполировать и адаптировать для ГКР-определения аналитов в реальных БЖЧ. В работах [80, 81] проводили несколько циклов электростатической сорбции/десорбции аналитов на поверхности ГКР-активного электрода. Показана возможность многократного использования таких ГКР-подложек, что особенно важно в случае дорогостоящих подложек на основе благородных металлов.
Жидкостно-жидкостная экстракция. Для определения гидрофобных соединений и снижения влияния компонентов БЖЧ на аналитический ГКР-сигнал предложено использовать предварительный этап ЖЖЭ. Например, разработаны методики ЖЖЭ-ГКР-определения некоторых НС (кокаин [56], метамфетамин [64, 65], морфин [53]), а также противоэпилептических и антибактериальных ЛС [19, 45]. В ряде работ по ЖЖЭ-ГКР-анализу использовали гидрофобные ГКР-подложки, позволяющие детектировать аналиты непосредственно в среде экстрагента сразу после экстракции аналита (без высушивания и перевода в водный раствор) [53, 56, 64, 65] и, как следствие, сократить продолжительность анализа. Применимость такого подхода показана при качественном определении морфина (вариант “да/нет”) в моче наркозависимых людей [53].
Описан пример использования жидкостной экстракции в нанесенном слое для экстракции бензадиазепеинов из мочи с последующим ГКР-определением элюированных аналитов [46]. Детектирование бензадиазипинов в смеси с некоторыми НС и ЛС показало отсутствие влияния примесей на определение целевого аналита.
Важно, что применение оптимизированного этапа ЖЖЭ позволяет снизить влияние мешающих веществ на аналитический сигнал аналитов без необходимости разработки специальных (и более трудоемких в изготовлении) ГКР-подложек [19, 45, 51]. Например, при ЖЖЭ-ГКР-определении сульфаметоксазола в моче [19] разбавление мочи, использование оптимального растворителя и корректировка рН среды позволили увеличить степень извлечения аналита, а также значительно снизить фоновый ГКР-сигнал мочи, при этом в качестве ГКР-подложки применяли коллоидные НЧ серебра, синтезированные по простой и экспрессной стандартной методике. Дополнительно эффективность и точность разработанной ЖЖЭ-ГКР-методики протестировали с использованием образцов мочи с переменным содержанием внутренних компонентов (собранных в разное время дня у здоровых добровольцев и пациентов с онкозаболеваниями). В работе [45] показано, что помимо увеличения эффективности ЖЖЭ за счет корректировки рН максимальный ГКР-сигнал бензодиазепинов достигается за счет использования хлорида магния в качестве агрегирующего агента для цитратных НЧ золота (по сравнению с хлоридами калия, натрия и кальция).
Следует отметить, что, несмотря на простоту и экономичность и экспрессность анализа (менее 30 мин), общим недостатком реализованных ЖЖЭ-ГКР-методик является использование органических растворителей, не отвечающих требованиям “зеленой химии”. Таким образом, перспективным направлением в данной области является адаптация методик экстракции с использованием, например, ПАВ или глубоких эвтектических растворителей.
Твердофазная экстракция (ТФЭ). Можно выделить двух- и одноэтапные методики, основанные на сочетании ТФЭ с ГКР, различающиеся наличием этапа элюирования. Двухэтапные ТФЭ-ГКР-методики включают стандартную процедуру ТФЭ и дальнейшее измерение ГКР-сигнала смеси раствора элюированного аналита с ГКР-подложкой. Например, такой подход применили для определения синтетического каннабиоида в слюне [61]. В качестве миниатюрной колонки для ТФЭ использовали наконечник для пипеток объемом 10 мкл, заполненный сорбентом С18. Разработанная методика позволила достичь предела обнаружения, соизмеримого с таковым для иммуноферментного метода определения каннабиноидов, а также сократить продолжительность анализа в три раза по сравнению с иммуноферментным анализом (менее 40 мин). В работе [39] в качестве сорбента использовали НЧ магнетита, покрытые полимером с молекулярными отпечатками, которые использовали для селективной экстракции антибиотика клоксациллина из плазмы крови. Эффективность разработанного сорбента доказана отсутствием его связывания с антибиотиками других классов, такими как доксициклин, спирамицин и гентамицин. Однако разработанная ТФЭ-ГКР-методика включает продолжительный по времени этап инкубирования плазмы крови с сорбентом (1 ч).
Одноэтапные ТФЭ-ГКР-методики основаны на использовании ГКР-активных сорбентов как для концентрирования, так и для ГКР-определения аналита. Важно отметить, что в случае ГКР-активных сорбентов устраняются проблемы, связанные с коллоидной и химической стабильностью встроенных НЧ [82], что существенно улучшает воспроизводимость сигнала и точность анализа. Регистрация спектров непосредственно с ГКР-активного сорбента дополнительно позволяет исключить этап элюирования и, как следствие, снизить потери аналита, отказаться от использования растворителей и сократить продолжительность анализа. Такие методики реализованы в двух форматах: статическом и динамическом. В первом вариранте ГКР-активный сорбент используют для заполнения колонки для ТФЭ [83]. В динамическом варианте микрочастицы сорбента диспергированы в анализируемом растворе и перед ГКР-определением их отделяют центрифугированием [18] или магнитной сепарацией (если сорбент имеет встроенные магнитные НЧ [31, 39, 84]).
Например, для реализации статического формата ТФЭ-ГКР и определения в слюне ЛС и НС предложено использование стеклянных капилляров, заполненных силикагелем со встроенными НЧ серебра или золота [23, 43, 44]. В указанных работах показана высокая эффективность отделения целевых аналитов от муцина слюны матрицей силикагеля, а продолжительность ТФЭ-ГКР-анализа составила около 10 мин. Важно отметить, что реализация ГКР-активных сорбентов в виде подобных капилляров или картриджей упрощает их интеграцию в проточную систему анализа, что позволит автоматизировать процесс определения и повысит его воспроизводимость [85].
Динамический формат ТФЭ-ГКР-анализа использовали для определения в моче противоопухолевого препарата метотрексата с применением ГКР-активного сорбента на основе оксида алюминия со встроенными НЧ серебра [18]. Однако матрица оксида алюминия является сильно светорассеивающей средой, не позволяющей регистрировать ГКР-сигнал в глубине сорбента. Для увеличения ГКР-сигнала предложено растворение матрицы сорбента добавлением раствора гидроксида натрия. С одной стороны, растворение матрицы позволяет высвободить НЧ серебра и молекулы аналита, а с другой стороны, проводить одновременную корректировку pH перед ГКР-измерениями (метотрексат обладает максимальным ГКР-сигналом в щелочной среде). В работе [18] дополнительно оценили влияние основных внутренних компонентов мочи (солей, мочевины, креатинина) и их смесей на степень сорбции метотрексата на сорбенте и его ГКР-сигнал.
Работа [31] посвящена ТФЭ-ГКР-детектированию цитостатического препарата тиогуанина в сыворотке крови с применением ГКР-активных альгинатных микрочастиц. Использование полимерной матрицы позволяет удалять белки сыворотки за счет отделения высокомолекулярных компонентов мезопористой структурой частиц сорбента, а также селективно адсорбировать аналиты по заряду благодаря сродству альгинатной матрицы к катионам.
Отметим, что модифицирование матрицы перечисленных ГКР-активных сорбентов заряженными и/или полярными фрагментами (−COOH, −NH2, Al3+ и т.д.) позволяет расширить области их применение благодаря увеличению селективности сорбентов по отношению к другим аналитам и/или исключению сорбции некоторых низкомолекулярных компонентов БЖЧ [86–89]. Для увеличения селективности ГКР-активных сорбентов также применяют технологию молекулярного импринтинга, что реализовано при определении ЛС и других веществ в воде и в таких сложных объектах анализа, как продукты питания [90]. Однако указанные ТФЭ-ГКР-методики пока не апробированы для определения ЛС или НС в реальных БЖЧ.
Неудерживающая твердофазная экстракция (НТФЭ). Для разделения фонообразующих компонентов БЖЧ и молекул аналита в ряде работ предложено использование НТФЭ, основанной на захвате и удерживании молекул мешающих соединений вместо молекул аналита. Например, для определения НС в образцах слюны и плазмы крови использовали ГКР-подложку в варианте ГКР-активной тест-полоски [54]. Такая полоска имеет ГКР-активную область, до которой доходят только молекулы аналита в результате вертикальной диффузии образца БЖЧ по нитроцеллюлозной мембране. В работе [49] поверх ГКР-активной поверхности расположили несколько нитроцеллюлозных мембран, при прохождении через которые происходит частичная очистка аналита от компонентов сыворотки крови перед достижением ГКР-активной области, что приводит к уменьшению фонового сигнала.
Для определения в моче цефалоспориновых антибиотиков предложены два варианта пробоподготовки, основанные на корректировке рН анализируемых растворов и НТФЭ некоторых фонообразующих компонентов мочи распространенными сорбентами [35, 36, 91]. Так, пропускание подщелоченного образца мочи через колонку с силикагелем позволило устранить сильный фоновый ГКР-сигнал мочи, обусловленный присутствием креатинина (рис. 4а) [35]. Авторы отметили, что варьирование величины pH и содержания белка в исходной моче в пределах значений, характерных для здоровых людей, не влияет на эффективность отделения аналита от матрицы мочи. В работе [36] для минимизации фонового сигнала мочи с помощью НТФЭ использовали гель гидроксида алюминия, который сорбирует уробилин мочи. В отличие от креатинина, уробилин имеет интенсивный ГКР-сигнал в кислой среде, что мешает ГКР-определению некоторых аналитов, также обладающих ГКР-сигналом в кислой среде (в том числе цефалоспоринов). Наконец, для устранения фонового ГКР-сигнала мочи, обусловленного присутствием мочевой кислоты, предложено ее удаление с помощью порошка оксида цинка (30 мас. %), хорошо адсорбирующего этот метаболит [91]. Благодаря такой пробоподготовке ГКР-сигнал мочевой кислоты полностью устраняется и появляется четкий ГКР-сигнал аналита (фенилаланин) (рис. 4б).
Рис. 4.
(a): Спектры гигантского комбинационного рассеяния образца мочи без и с добавкой цефтриаксона (ЦТР) до и после пропускания через колонку с силикагелем. Адаптировано из работы [35]. (б): Спектры гигантского комбинационного рассеяния эквимолярной (50 мкМ) смеси мочевой кислоты (МК) и фенилаланина (ФА) до и после контакта с порошком оксида цинка (2 мас. %), а также спектры МК и ФА в отдельности. Адаптировано из статьи [91] с разрешения правобладателя.

Таким образом, удаление мешающих компонентов с помощью НТФЭ позволяет увеличить точность ГКР-определения ЛС и НС в БЖЧ без использования органических растворителей и специальных сорбентов. Стоит отметить, что использование этапа НТФЭ незначительно усложняет анализ и во всех перечисленных работах продолжительность анализа не превышала 10 мин из-за малого числа манипуляций, а в качестве ГКР-подложек применяли НЧ металлов, синтезированные по известным и простым методикам. Однако важно помнить, что в ходе НТФЭ может происходить частичная сорбция молекул аналита и снижение интенсивности аналитического сигнала. Тем не менее в рассмотренных работах показано, что высокая чувствительность спектроскопии ГКР позволяет нивелировать этот недостаток и получать аналитический сигнал с интенсивностью, достаточной для определения ЛС и НС в необходимом диапазоне концентраций.
ЭФФЕКТИВНОСТЬ ПРИМЕНЕНИЯ СПЕКТРОСКОПИИ ГИГАНТСКОГО КОМБИНАЦИОННОГО РАССЕЯНИЯ В АНАЛИЗЕ
К сожалению, несмотря на большое число публикаций по применению ГКР для определения ЛС и НС в БЖЧ в журналах по аналитической химии (табл. 1), сделать однозначные выводы об эффективности и практической применимости предложенных методик пока затруднительно. Например, зачастую в публикациях отсутствует анализ соответствия определяемых концентраций практическим потребностям. В основном авторы просто пытаются достичь как можно более низких пределов обнаружения и широких рабочих диапазонов концентраций, игнорируя другие важные моменты. Так, определяемые концентрации ЛС в БЖЧ при проведении ТЛМ могут иметь достаточно высокие значения (особенно в моче), что объясняется необходимостью достижения терапевтического эффекта от применения ЛС. Также ЛС может иметь узкое окно терапевтических концентраций в пределах одного порядка. В данных случаях требования к высокой селективности и точности выходят на первое место.
Другим важным моментом, выявленным при оценке метрологических характеристик методик ГКР-анализа, является не вполне корректная обработка результатов анализа. Этот факт затрудняет сравнение эффективности различных методик, использованных для определения одного и того же аналита. Например, только в половине исследований, приведенных в таблице (табл. 1), построены градуировочные графики и корректно рассчитаны значения пределов обнаружения. Вместо предела обнаружения, рассчитанного согласно рекомендациям ИЮПАК, часто используют значения минимальной концентрации на градуировочном графике без учета погрешности измерения сигнала. Наконец, ощущается нехватка работ, содержащих результаты проверки предложенных ГКР-методик с привлечением контрольных методов.
К достоинствам рассмотренных методик ГКР-анализа можно отнести активное использование портативных КР-спектрометров (примерно в трети работ). Известно, что такие спектрометры имеют более низкую чувствительность по сравнению со стационарными КР-микроскопами, в основном используемыми в фундаментальных научных исследованиях. Этот факт дополнительно подтверждает возможность проведения экспрессного ГКР-анализа на месте.
Наконец, самым важным достижением является достаточно широкое тестирование методик ГКР-анализа с использованием клинических образцов БЖЧ, полученных от волонтеров, принимавших ЛС или НС (табл. 1). Таким образом, несмотря на отсутствие детального изучения селективности и влияющих на нее факторов, проведенные исследования показывают, что ГКР пригоден для анализа реальных образцов БЖЧ.
* * *
Подводя итоги, можно сделать вывод, что в настоящее время основные усилия при разработке методик ГКР-определения ЛС и НС в БЖЧ в первую очередь направлены на улучшение селективности и подавление фонового сигнала. Для этого разрабатывают ГКР-методики без и с привлечения методов разделения и концентрирования. К первым относятся методики с простыми способами пробоподготовки (разбавление и варьирование рН) и с обработкой ГКР-сигнала методами хемометрики. Сюда же можно условно отнести использование модификации поверхности ГКР-подложек для улучшения сорбционных свойств и селективности (мультифункциональных ГКР-подложек). Во втором случае ГКР-методики включают предварительное отделение молекул аналита от примесей с использованием экстракции (удерживающая и неудерживающая ТФЭ, ЖЖЭ) и хроматографии (ТСХ, ВЭЖХ), а также электрофоретического разделения. Анализ метрологических характеристик опубликованных методик показал, что, несмотря на положительные результаты при работе с реальными образцами БЖЧ и существенный прогресс в создании эффективных ГКР-подложек, необходимы дальнейшие исследования. На наш взгляд, перспективным направлением является создание автоматизированных систем ГКР-анализа с высокой скоростью сбора и обработки данных, что важно для внедрения ГКР в клиническую диагностику.
Работа выполнена при финансовой поддержке Российского научного фонда (проект № 21-73-00098).
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Burke M.D. Principles of therapeutic drug monitoring // Postgrad. Med. 1981. V. 70. P. 57. https://doi.org/10.1080/00325481.1981.11715801
Ensom M.H.H., Davis G.A., Cropp C.D., Ensom R.J. Clinical pharmacokinetics in the 21st century // Clin. Pharmacokinet. 1998. V. 34. P. 265. https://doi.org/10.2165/00003088-199834040-00001
Vogeser M., Seger C. A decade of HPLC–MS/MS in the routine clinical laboratory – Goals for further developments // Clin. Biochem. 2008. V. 41. P. 649. https://doi.org/10.1016/j.clinbiochem.2008.02.017
Brandhorst G., Oellerich M., Maine G., Taylor P., Veen G., Wallemacq P. Liquid chromatography–tandem mass spectrometry or automated immunoassays: What are the future trends in therapeutic drug monitoring? // Clin. Chem. 2012. V. 58. P. 821. https://doi.org/10.1373/clinchem.2011.167189
Shipkova M., Petrova D.T., Rosler A.E., Orth M., Engelmayer J., Wieland E., Brandhorst G., Oellerich M. Comparability and imprecision of 8 frequently used commercially available immunoassays for therapeutic drug monitoring // Ther. Drug Monit. 2014. V. 36. P. 433. https://doi.org/10.1097/FTD.0000000000000043
Jaworska A., Fornasaro S., Sergo V., Bonifacio A. Potential of surface enhanced Raman spectroscopy (SERS) in therapeutic drug monitoring (TDM). A critical review // Biosensors. 2016. V. 6. Article 47. https://doi.org/10.3390/bios6030047
Yu B., Ge M., Li P., Xie Q., Yang L. Development of surface-enhanced Raman spectroscopy application for determination of illicit drugs: Towards a practical sensor // Talanta. 2019. V. 191. P. 1. https://doi.org/10.1016/j.talanta.2018.08.032
Li C., Ye Z., Xu Y., Bell S.E.J. An overview of therapeutic anticancer drug monitoring based on surface enhanced (resonance) Raman spectroscopy (SE(R)RS) // Analyst. 2020. V. 145. P. 6211. https://doi.org/10.1039/D0AN00891E
Jahn M., Patze S., Hidi I.J., Knipper R., Radu A.I., Mühlig A., Yüksel S., Peksa V., Weber K., Mayerhöfer T., Cialla-May D., Popp J. Plasmonic nanostructures for surface enhanced spectroscopic methods // Analyst. 2016. V. 141. P. 756. https://doi.org/10.1039/C5AN02057C
Bonifacio A., Marta S.D., Spizzo R., Cervo S., Steffan A., Colombatti A., Sergo V. Surface-enhanced Raman spectroscopy of blood plasma and serum using Ag and Au nanoparticles: A systematic study // Anal. Bioanal. Chem. 2014. V. 406. P. 2355. https://doi.org/10.1007/s00216-014-7622-1
Fornasaro S., Alsamad F., Baia M., Batista de Carvalho L.A.E., Beleites C., Byrne H.J., Chiadò A., Chis M., Chisanga M., Daniel A., Dybas J., Eppe G., Falgayrac G., Faulds K., Gebavi H., Giorgis F., Goodacre R., Graham D., La Manna P., Laing S., Litti L., Lyng F.M., Malek K., Malherbe C., Marques M.P.M., Meneghetti M., Mitri E., Mohaček-Grošev V., Morasso C., Muhamadali H., Musto P., Novara C., Pannico M., Penel G., Piot O., Rindzevicius T., Rusu E.A., Schmidt M.S., Sergo V., Sockalingum G.D, Untereiner V., Vanna R., Wiercigroch E., Bonifacio A. Surface enhanced Raman spectroscopy for quantitative analysis: Results of a large-scale European multi-instrument interlaboratory study // Anal. Chem. 2020. V. 92. P. 4053. https://doi.org/10.1021/acs.analchem.9b05658
Guo S., Beleites C., Neugebauer U., Abalde-Cela S., Afseth N.K., Alsamad F., Anand S., Araujo-Andrade C., Aškrabić S., Avci E., Baia M., Baranska M., Baria E., Batista de Carvalho L.A.E., de Bettignies P., Bonifacio A., Bonnier F., Brauchle E.M., Byrne H.J., Chourpa I., Cicchi R., Cuisinier F., Culha M., Dahms M., David C., Duponchel L., Duraipandian S., El-Mashtoly S.F., Ellis D.I., Eppe G., Falgayrac G., Gamulin O., Gardner B., Gardner P., Gerwert K., Giamarellos-Bourboulis E.J., Gizurarson S., Gnyba M., Goodacre R., Grysan P., Guntinas-Lichius O., Helgadottir H., Mohaček Grošev V., Kendall C., Kiselev R., Kölbach M., Krafft C., Krishnamoorthy S., Kubryck P., Lendl B., Loza-Alvarez P., Lyng F.M., Machill S., Malherbe C., Marro M., Marques M.P.M., Matuszyk E., Morasso C.F., Moreau M., Muhamadali H., Mussi V., Notingher I., Pacia M.Z., Pavone F.S., Penel G., Petersen D., Piot O., Rau J.V., Richter M., Rybarczyk M.K., Salehi H., Schenke-Layland K., Schlücker S., Schosserer M., Schütze K., Sergo V., Sinjab F., Smulko J., Sockalingum G.D., Stiebing C., Stone N., Untereiner V., Vanna R., Wieland K., Popp J., Bocklitz T. Comparability of Raman spectroscopic configurations: A large scale cross-laboratory study // Anal. Chem. 2020. V. 92. P. 15745. https://doi.org/10.1021/acs.analchem.0c02696
Bell S.E.J., Sirimuthu N.M.S. Quantitative surface-enhanced Raman spectroscopy // Chem. Soc. Rev. 2008. V. 37. P. 1012. https://doi.org/10.1039/B705965P
Fang Y., Seong N.H., Dlott D.D. Measurement of the distribution of site enhancements in surface-enhanced Raman scattering // Science. 2008. V. 321. P. 388. https://doi.org/10.1126/science.1159499
Goodacre R., Graham D., Faulds K. Recent developments in quantitative SERS moving towards absolute quantification // Trends Anal. Chem. 2018. V. 102. P. 359. https://doi.org/10.1016/j.trac.2018.03.005
Zhu Q.,Yu X., Wu Z., Lu F., Yuan Y. Antipsychotic drug poisoning monitoring of clozapine in urine by using coffee ring effect based surface-enhanced Raman spectroscopy // Anal. Chim. Acta. 2018. V. 1014. P. 64. https://doi.org/10.1016/j.aca.2018.02.027
Markina N.E., Ustinov S.N., Zakharevich A.M., Markin A.V. Copper nanoparticles for SERS-based determination of some cephalosporin antibiotics in spiked human urine // Anal. Chim. Acta. 2020. V. 1138. P. 9. https://doi.org/10.1016/j.aca.2020.09.016
Markina N.E., Zakharevich A.M., Markin A.V. Determination of methotrexate in spiked human urine using SERS-active sorbent // Anal. Bioanal. Chem. 2020. V. 412. P. 7757. https://doi.org/10.1007/s00216-020-02932-x
Markina N.E., Markin A.V., Weber K., Popp J., Cialla-May D. Liquid-liquid extraction-assisted SERS determination of sulfamethoxazole in spiked human urine // Anal. Chim. Acta. 2020. V. 1109. P. 61. https://doi.org/10.1016/j.aca.2020.02.067
Markin A.V., Markina N.E., Popp J., Cialla-May D. Copper nanostructures for chemical analysis using surface-enhanced Raman spectroscopy // Trends Anal. Chem. 2018. V. 108, P. 247. https://doi.org/10.1016/j.trac.2018.09.004
Beljebbar A., Sockalingum G.D., Angiboust J.F., Manfait M. Comparative FT SERS, resonance Raman and SERRS studies of doxorubicin and its complex with DNA // Spectrochim. Acta A. 1995. V. 51. P. 2083. https://doi.org/10.1016/0584-8539(95)01515-7
McLaughlin C., MacMillan D., McCardle C., Smith W.E. Quantitative analysis of mitoxantrone by surface-enhanced resonance Raman scattering // Anal. Chem. 2002. V. 74. P. 3160. https://doi.org/10.1021/ac010067k
Farquharson S., Gift A.D., Shende C., Maksymiuk P., Inscore F.E., Murran J. Detection of 5-fluorouracil in saliva using surface-enhanced Raman spectroscopy // Vib. Spectrosc. 2005. V. 38. P. 79. https://doi.org/10.1016/j.vibspec.2005.02.021
Farquharson S., Gift A., Shende C., Inscore F., Ordway B., Farquharson C., Murren J. Surface-enhanced Raman spectral measurements of 5-fluorouracil in saliva // Molecules. 2008. V. 13. P. 2608. https://doi.org/10.3390/molecules13102608
Yuen C., Zheng W., Huang Z. Low-level detection of anti-cancer drug in blood plasma using microwave-treated gold-polystyrene beads as surface-enhanced Raman scattering substrates // Biosens. Bioelectron. 2010. V. 26. P. 580. https://doi.org/10.1016/j.bios.2010.07.030
Dong J., Chen Q., Rong C., Li D., Rao Y. Minimally invasive surface-enhanced Raman scattering detection with depth profiles based on a surface-enhanced Raman scattering-active acupuncture needle // Anal. Chem. 2011. V. 83. P. 6191. https://doi.org/10.1021/ac2007009
Fornasaro S., Marta S.D., Rabusin M., Bonifacio A., Sergo V. Toward SERS-based point-of-care approaches for therapeutic drug monitoring: The case of methotrexate // Faraday Discuss. 2016. V. 187. P. 485. https://doi.org/10.1039/C5FD00173K
Sun F., Hung H.C., Sinclair A., Zhang P., Bai T., Galvan D.D., Jain P., Li B., Jiang S., Yu Q. Hierarchical zwitterionic modification of a SERS substrate enables real-time drug monitoring in blood plasma // Nat. Commun. 2016. V. 7. Article 13437. https://doi.org/10.1038/ncomms13437
Subaihi A., Trivedi D.K., Hollywood K.A., Bluett J., Xu Y., Muhamadali H., Ellis D.I., Goodacre R. Quantitative online liquid chromatography−surface-enhanced Raman scattering (LC-SERS) of methotrexate and its major metabolites // Anal. Chem. 2017. V. 89. P. 6702. https://doi.org/10.1021/acs.analchem.7b00916
Fornasaro S., Bonifacio A., Marangon E., Buzzo M., Toffoli G., Rindzevicius T., Schmidt M.S., Sergo V. Label-free quantification of anticancer drug imatinib in human plasma with surface enhanced Raman spectroscopy // Anal. Chem. 2018. V. 90. P. 12670. https://doi.org/10.1021/acs.analchem.8b02901
Yue S., Sun X.T., Wang Y., Zhang W.S., Xu Z.R. Microparticles with size/charge selectivity and pH response for SERS monitoring of 6-thioguanine in blood serum // Sens. Actuators B. 2018. V. 273. P. 1539. https://doi.org/10.1016/j.snb.2018.07.062
Litti L., Ramundo A., Biscaglia F., Toffoli G., Gobbo M., Meneghetti M. A surface enhanced Raman scattering based colloid nanosensor for developing therapeutic drug monitoring // J. Colloid Interface Sci. 2019. V. 533. P. 621. https://doi.org/10.1016/j.jcis.2018.08.107
Panikar S.S., Ramírez-García G., Sidhik S., Lopez-Luke T., Rodriguez-Gonzalez C., Ciapara I.H., Castillo P.S., Camacho-Villegas T., De la Rosa E. Ultrasensitive SERS substrate for label-free therapeutic-drug monitoring of paclitaxel and cyclophosphamide in blood serum // Anal. Chem. 2019. V. 91. P. 2100. https://doi.org/10.1021/acs.analchem.8b04523
Zhang W.S., Wang Y.N., Wang Y., Xu Z.R. Highly reproducible and fast detection of 6-thioguanine in human serum using a droplet-based microfluidic SERS system // Sens. Actuators B. 2019. V. 283. P. 532. https://doi.org/10.1016/j.snb.2018.12.077
Markina N.E., Goryacheva I.Yu., Markin A.V. Sample pretreatment and SERS-based detection of ceftriaxone in urine // Anal. Bioanal. Chem. 2018. V. 410. P. 2221. https://doi.org/10.1007/s00216-018-0888-y
Markina N.E., Markin A.V. Application of aluminum hydroxide for improvement of label-free SERS detection of some cephalosporin antibiotics in urine // Biosensors. 2019. V. 9. Article 91. https://doi.org/10.3390/bios9030091
Mamián-López M.B., Poppi R.J. Quantification of moxifloxacin in urine using surface-enhanced Raman spectroscopy (SERS) and multivariate curve resolution on a nanostructured gold surface // Anal. Bioanal. Chem. 2013. V. 405. P. 7671. https://doi.org/10.1007/s00216-013-7200-y
Hidi I.J., Jahn M., Pletz M.W., Weber K., Cialla-May D., Popp J. Toward levofloxacin monitoring in human urine samples by employing the LoC-SERS technique // J. Phys. Chem. C. 2016. V. 120. P. 20613. https://doi.org/10.1021/acs.jpcc.6b01005
Ashley J., Wu K., Hansen M.F., Schmidt M.S., Boisen A., Sun Y. Quantitative detection of trace level cloxacillin in food samples using magnetic molecularly imprinted polymer extraction and surface-enhanced Raman spectroscopy nanopillars // Anal. Chem. 2017. V. 89. P. 11484. https://doi.org/10.1021/acs.analchem.7b02725
Liu J., Liu W., Huang Y., Zhao X., Feng Z., Wang D., Gong Z., Fan M. Self-supporting liquid film as reproducible SERS platform for therapeutic drug monitoring of berberine hydrochloride in human urine // Microchem. J. 2021. V. 165. Article 106122. https://doi.org/10.1016/j.microc.2021.106122
Hidi I.J., Jahn M., Weber K., Bocklitz T., Pletz M.W., Cialla-May D., Popp J. Lab-on-a-chip-surface enhanced Raman scattering combined with the standard addition method: Toward the quantification of nitroxoline in spiked human urine samples // Anal. Chem. 2016. V. 88. P. 9173. https://doi.org/10.1021/acs.analchem.6b02316
Trachta G., Schwarze B., Sagmuller B., Brehm G., Schneidera S. Combination of high-performance liquid chromatography and SERS detection applied to the analysis of drugs in human blood and urine // J. Mol. Struct. 2004. V. 693. P. 175. https://doi.org/10.1016/j.molstruc.2004.02.034
Farquharson S., Shende C., Sengupta A., Huang H., Inscore F. Rapid detection and identification of overdose drugs in saliva by surface-enhanced Raman scattering using fused gold colloids // Pharmaceutics. 2011. V. 3. P. 425. https://doi.org/10.3390/pharmaceutics3030425
Inscore F., Shende C., Sengupta A., Huang H., Farquharson S. Detection of drugs of abuse in saliva by surface-enhanced Raman spectroscopy (SERS) // Appl. Spectrosc. 2011. V. 65. P. 1004. https://doi.org/10.1366/11-06310
Doctor E.L., McCord B. Comparison of aggregating agents for the surface-enhanced Raman analysis of benzodiazepines // Analyst. 2013. V. 138. P. 5926. https://doi.org/10.1039/C3AN00669G
Doctor E.L., McCord B. The application of supported liquid extraction in the analysis of benzodiazepines using surface enhanced Raman spectroscopy // Talanta. 2015. V. 144. P. 938. https://doi.org/10.1016/j.talanta.2015.07.036
Lucotti A., Tommasini M., Casella M., Morganti A., Gramatica F., Zerbi G. TLC–surface enhanced Raman scattering of apomorphine in human plasma // Vib. Spectrosc. 2012. V. 62. P. 286. https://doi.org/10.1016/j.vibspec.2012.07.009
Phung V.D., Jung W.S., Nguyen T.A., Kim J.H., Lee S.W. Reliable and quantitative SERS detection of dopamine levels in human blood plasma using a plasmonic Au/Ag nanocluster substrate // Nanoscale. 2018. V. 10. P. 22493. https://doi.org/10.1039/C8NR06444J
Berger A.G., Restaino S.M., White I.M. Vertical-flow paper SERS system for therapeutic drug monitoring of flucytosine in serum // Anal. Chim. Acta. 2017. V. 949. P. 59. https://doi.org/10.1016/j.aca.2016.10.035
Gan S., Shi X., Zhu X., Wu C., Li Z., Han T., Lu R. Rapid dynamic determination of cetirizine dihydrochloride in urine using surface enhanced Raman scattering with silver colloids // Anal. Lett. 2018. V. 51. P. 1163. https://doi.org/10.1080/00032719.2017.1370597
Cheng J., Wang S., Zhang S., Wang P., Xie J., Han C., Su X.O. Rapid and sensitive determination of clenbuterol residues in animal urine by surface-enhanced Raman spectroscopy // Sens. Actuators B. 2019. V. 279. P. 7. https://doi.org/10.1016/j.snb.2018.09.103
Subaihi A., Muhamadali H., Mutter S.T., Blanch E., Ellisa D.I., Goodacre R. Quantitative detection of codeine in human plasma using surface-enhanced Raman scattering via adaptation of the isotopic labelling principle // Analyst. 2017. V. 142. P. 1099. https://doi.org/10.1039/C7AN00193B
Yu B., Cao C., Li P., Mao M., Xie Q., Yang L. Sensitive and simple determination of zwitterionic morphine in human urine based on liquid-liquid micro-extraction coupled with surface-enhanced Raman spectroscopy // Talanta. 2018. V. 186. P. 427. https://doi.org/10.1016/j.talanta.2018.04.094
Shende C., Brouillette C., Farquharson S. Detection of codeine and fentanyl in saliva, blood plasma and whole blood in 5-minutes using a SERS flow-separation strip // Analyst. 2019. V. 144. P. 5449. https://doi.org/10.1039/C9AN01087D
Huang R., Han S., Li X. Detection of tobacco-related biomarkers in urine samples by surface-enhanced Raman spectroscopy coupled with thin-layer chromatography // Anal. Bioanal. Chem. 2013. V. 405. P. 6815. https://doi.org/10.1007/s00216-013-7107-7
Meng J., Tang X., Zhou B., Xie Q., Yang L. Designing of ordered two-dimensional gold nanoparticles film for cocaine detection in human urine using surface-enhanced Raman spectroscopy // Talanta. 2017. V. 164. P. 693. https://doi.org/10.1016/j.talanta.2016.10.101
Dies H., Raveendran J., Escobedo C., Docoslis A. Rapid identification and quantification of illicit drugs on nanodendritic surface-enhanced Raman scattering substrates // Sens. Actuators B. 2018. V. 257. P. 382. https://doi.org/10.1016/j.snb.2017.10.181
Kong X., Chong X., Squire K., Wang A.X. Microfluidic diatomite analytical devices for illicit drug sensing with ppb-level sensitivity // Sens. Actuators B. 2018. V. 259. P. 587. https://doi.org/10.1016/j.snb.2017.12.038
Sivashanmugan K., Zhao Y., Wang A.X. Tetrahydrocannabinol sensing in complex biofluid with portable Raman spectrometer using diatomaceous SERS substrates // Biosensors. 2019. V. 9. Article 125. https://doi.org/10.3390/bios9040125
Alder R., Xiao L., Fu S. Comparison of commercial surface-enhanced Raman spectroscopy substrates for the analysis of cocaine // Drug Test. Anal. 2021. V. 13. P. 944. https://doi.org/10.1002/dta.2894
Deriu C., Conticello I., Mebel A.M., McCord B. Micro solid phase extraction surface-enhanced Raman spectroscopy (μ-SPE/SERS) screening test for the detection of the synthetic cannabinoid JWH-018 in oral fluid // Anal. Chem. 2019. V. 91. P. 4780. https://doi.org/10.1021/acs.analchem.9b00335
Andreou C., Hoonejani M.R., Barmi M.R., Moskovits M., Meinhart C.D. Rapid detection of drugs of abuse in saliva using surface enhanced Raman spectroscopy and microfluidics // ACS Nano. 2013. V. 7. P. 7157. https://doi.org/10.1021/nn402563f
Dong R., Weng S., Yang L., Liu J. Detection and direct readout of drugs in human urine using dynamic surface-enhanced Raman spectroscopy and support vector machines // Anal. Chem. 2015. V. 87. P. 2937. https://doi.org/10.1021/acs.analchem.5b00137
Han Z., Liu H., Meng J., Yang J., Liu J., Liu J. Portable kit for identification and detection of drugs in human urine using surface-enhanced Raman spectroscopy // Anal. Chem. 2015. V. 87. P. 9500. https://doi.org/10.1021/acs.analchem.5b02899
Han Z., Liu H., Wang B., Weng S., Yang L., Liu J. Three-dimensional surface-enhanced Raman scattering hotspots in spherical colloidal superstructure for identification and detection of drugs in human urine // Anal. Chem. 2015. V. 87. P. 4821. https://doi.org/10.1021/acs.analchem.5b00176
Nuntawong N., Eiamchai P., Somrang W., Denchitcharoen S., Limwichean S., Horprathum M., Patthanasettakul V., Chaiya S., Leelapojanaporn A., Saiseng S., Pongsethasant P., Chindaudom P. Detection of methamphetamine/amphetamine in human urine based on surface-enhanced Raman spectroscopy and acidulation treatments // Sens. Actuators B. 2017. V. 239. P. 139. https://doi.org/10.1016/j.snb.2016.07.129
Velička M., Zacharovas E., Adomavičiūtė S., Šablinskas V. Detection of caffeine intake by means of EC-SERS spectroscopy of human saliva // Spectrochim. Acta A. 2021. V. 246. Article 118956. https://doi.org/10.1016/j.saa.2020.118956
Fan M., Andrade G.F.S., Brolo A.G. A review on the fabrication of substrates for surface enhanced Raman spectroscopy and their applications in analytical chemistry // Anal. Chim. Acta. 2011. V. 693. P. 7. https://doi.org/10.1016/j.aca.2011.03.002
Zhang Y., Li L., Gao Y., Wang X., Sun L., Ji W., Ozaki Y. Nitrosonaphthol reaction-assisted SERS assay for selective determination of 5-hydroxyindole-3-acetic acid in human urine // Anal. Chim. Acta. 2020. V. 1134. P. 34. https://doi.org/10.1016/j.aca.2020.08.020
Yang H., Xiang Y., Guo X., Wu Y., Wen Y., Yang H. Diazo-reaction-based SERS substrates for detection of nitrite in saliva // Sens. Actuators B. 2018. V. 271. P. 118. https://doi.org/10.1016/j.snb.2018.05.111
Cao G., Hajisalem G., Li W., Hof F., Gordon R. Quantification of an exogenous cancer biomarker in urinalysis by Raman Spectroscopy // Analyst. 2014. V. 139. P. 5375. https://doi.org/10.1039/C4AN01309C
Markina N.E., Cialla-May D., Markin A.V. Cyclodextrin-assisted surface-enhanced Raman spectroscopy: A critical review // Anal. Bioanal. Chem. 2022. V. 414. P. 923. https://doi.org/10.1007/s00216-021-03704-x
Kleinman S.L., Frontiera R.R., Henry A.I., Dieringer J.A., Van Duyne R.P. Creating, characterizing, and controlling chemistry with SERS hot spots // Phys. Chem. Chem. Phys. 2013. V. 15. P. 21. https://doi.org/10.1039/C2CP42598J
Gao F., Hu Y., Chen D., Li-Chan E.C.Y., Grant E., Lu X. Determination of Sudan I in paprika powder by molecularly imprinted polymers–thin layer chromatography–surface enhanced Raman spectroscopic biosensor // Talanta. 2015. V. 143. P. 344. https://doi.org/10.1016/j.talanta.2015.05.003
bin Zhang B., Shi Y., Chen H., xia Zhu Q., Lu F., wei Li Y. A separable surface-enhanced Raman scattering substrate modified with MIL-101 for detection of overlapping and invisible compounds after thin-layer chromatography development // Anal. Chim. Acta. 2018. V. 997. P. 35. https://doi.org/10.1016/j.aca.2017.10.006
Lee B.S., Lin P.C., Lin D.Z., Yen T.J. Rapid biochemical mixture screening by three-dimensional patterned multifunctional substrate with ultra-thin layer chromatography (UTLC) and surface enhanced Raman scattering (SERS) // Sci. Rep. 2018. V. 8. Article 516. https://doi.org/10.1038/s41598-017-18967-7
Li Y.T., Qu L.L., Lin D.W., Song Q.X., Fathi F., Long Y.T. Rapid andsensitive in-situ detection of polar antibiotics in water using a disposable Ag–graphene sensor based on electrophoretic preconcentration and surface-enhanced Raman spectroscopy // Biosens. Bioelectron. 2013. V. 43. P. 94. https://doi.org/10.1016/j.bios.2012.12.005
Bindesri S.D., Alhatab D.S., Brosseau C.L. Development of an electrochemical surface-enhanced Raman spectroscopy (EC-SERS) fabric-based plasmonic sensor for point-of-care diagnostics // Analyst. 2018. V. 143. P. 4128. https://doi.org/10.1039/C8AN01117F
Bindesri S.D., Jebailey R., Albarghouthi N., Pye C.C., Brosseau C.L. Spectroelectrochemical and computational studies of tetrahydrocannabinol (THC) and carboxy-tetrahydrocannabinol (THC-COOH) // Analyst. 2020. V. 145. P. 1849. https://doi.org/10.1039/C9AN02173F
Höhn E.M., Panneerselvam R., Das A., Belder D. Raman spectroscopic detection in continuous microflow using a chip-integrated silver electrode as an electrically regenerable surface-enhanced Raman spectroscopy substrate // Anal. Chem. 2019. V. 91. P. 9844. https://doi.org/10.1021/acs.analchem.9b01514
Viehrig M., Rajendran S.T., Sanger K., Schmidt M.S., Alstrøm T.S., Rindzevicius T., Zór K., Boisen A. Quantitative SERS assay on a single chip enabled by electrochemically assisted regeneration: A method for detection of melamine in milk // Anal. Chem. 2020. V. 92. P. 4317. https://doi.org/10.1021/acs.analchem.9b05060
Markina N.E., Volkova E.K., Zakharevich A.M., Goryacheva I.Yu., Markin A.V. SERS detection of ceftriaxone and sulfadimethoxine using copper nanoparticles temporally protected by porous calcium carbonate // Microchim. Acta. 2018. V. 185. Article 481. https://doi.org/10.1007/s00604-018-3018-9
Markina N.E., Markin A.V., Zakharevich A.M., Gorin D.A., Rusanova T.Yu., Goryacheva I.Yu. Multifunctional silver nanoparticle-doped silica for solid-phase extraction and surface-enhanced Raman scattering detection // J. Nanopart. Res. 2016. V. 18. Article 353. https://doi.org/10.1007/s11051-016-3661-1
Markina N.E., Markin A.V., Zakharevich A.M., Goryacheva I.Yu. Calcium carbonate microparticles with embedded silver and magnetite nanoparticles as new SERS-active sorbent for solid phase extraction // Microchim. Acta. 2017. V. 184. P. 3937. https://doi.org/10.1007/s00604-017-2426-6
Jahn I.J., Zukovskaja O., Zheng X.S., Weber K., Bocklitz T.W., Cialla-May D., Popp J. Surface-enhanced Raman spectroscopy and microfluidic platforms: Challenges, solutions and potential applications // Analyst. 2017. V. 142. P. 1022. https://doi.org/10.1039/C7AN00118E
Marsich L., Bonifacio A., Mandal S., Krol S., Beleites C., Sergo V. Poly-l-lysine-coated silver nanoparticles as positively charged substrates for surface-enhanced Raman scattering // Langmuir. 2012. V. 28. P. 13166. https://doi.org/10.1021/la302383r
López-Puente V., Angelomé P.C., Soler-Illia G.J.A.A., Liz-Marzán L.M. Selective SERS sensing modulated by functionalized mesoporous films // ACS Appl. Mater. Interfaces. 2015. V. 7. P. 25633. https://doi.org/10.1021/acsami.5b10543
Yurova N.S., Markina N.E., Pozharov M.V., Zakhare-vich A.M., Rusanova T.Yu., Markin A.V. SERS-active sorbent based on aluminum oxide loaded with silver nanoparticles // Colloids Surf. A. 2016. V. 495. P. 169. https://doi.org/10.1016/j.colsurfa.2016.02.006
Markina N.E., Zakharevich A.M., Markin A.V. Improvement of creatinine SERS detection using molecularly imprinted silica gel // Proc. SPIE. 2019. V. 11065. Article 110651M. https://doi.org/10.1117/12.2521212
Guo X., Li J., Arabi M., Wang X., Wang Y., Chen L. Molecular-imprinting-based surface-enhanced Raman scattering sensors // ACS Sens. 2020. V. 5. P. 601. https://doi.org/10.1021/acssensors.9b02039
Murugesan B., Yang J. Tunable coffee ring formation on polycarbonate nanofiber film for sensitive SERS detection of phenylalanine in urine // ACS Omega. 2019. V. 4. P. 14928. https://doi.org/10.1021/acsomega.9b01686
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии